
Abstract
The MAST-4 (marine stramenopile group 4) is a widespread uncultured picoeukaryote that makes up an important fraction of marine heterotrophic flagellates. This group has low genetic divergence and is composed of a small number of putative species. We combined ARISA (automated ribosomal intergenic spacer analysis) and ITS (Internal Transcribed Spacer) clone libraries to study the biogeography of this marine protist, examining both spatial and temporal trends in MAST-4 assemblages and associated environmental factors. The most represented MAST-4 clades appeared adapted to different temperature ranges, and their distributions did not suggest clear geographical barriers for dispersal. Distant samples sharing the same temperature had very similar assemblages, especially in cold temperatures, where only one clade, E1, dominated. The most highly represented clades, A and E1, showed very little differentiation between populations from distant geographical regions. Within a single site, temporal variation also followed patterns governed by temperature. Our results contribute to the general discussion on microbial biogeography by showing strong environmental selection for some picoeukaryotes in the marine environment.
Similar content being viewed by others


Introduction
Biogeography is the study of the distribution of biodiversity over space and time. Studies performed with plants and animals have revealed geographically restricted metapopulations due to deterministic (environmental) and stochastic (random dispersal) processes (MacArthur and Wilson, 1967). Whether or not microorganisms follow similar patterns is an old debate (Beijerinck, 1913) that has recently seen a renovated interest (O’Malley, 2008, Hanson et al., 2012). A long-held concept is that free-living organisms <1 mm (all prokaryotes and most protists) are sufficiently abundant to have worldwide distribution owing to their dispersal ability (Finlay, 2002, Fenchel and Finlay, 2004). Microbial cells can be dispersed via wind, water or birds, and many species have a high ability to become dormant in harsh environments (Whitfield, 2005). Consequently, microbial organisms are believed to occur wherever the environment permits: ‘everything is everywhere, but the environment selects’ (Baas-Becking, 1934). Implicit in this tenet is that free-living microbial taxa are not randomly distributed but exhibit biogeographical patterns, and in some cases, these patterns may be qualitatively similar to those observed for macroorganisms (Green et al., 2004, Horner-Devine et al., 2007). However, as indicated in the tenet, these patterns would be the result of local environmental selection rather than dispersal limitation.
The current evidence confirms that environmental selection is fundamental for the spatial variation observed in microbial diversity (Martiny et al., 2006). The next frontier is to figure out whether these patterns are also influenced by geographical barriers that facilitate evolution and diversification. Because geographical distance is often correlated with specific environmental characteristics, disentangling the relative influence of these two factors on community divergence represents a major challenge in elucidating whether or not microbes are limited by dispersal. It is known that freshwater diatoms present dispersal limitations because of desiccation intolerances (Vyverman et al., 2007). Other highly specialized microorganisms, such as the hyperthermophiles, are unlikely to make a long dispersal journey, so are easily isolated by geographic barriers, resulting in the development of a global population structure (Whitaker, 2003). However, marine systems are expected to be less prone to dispersal limitation than terrestrial and freshwater systems.
The oceans are an interconnected geophysical fluid that potentially allows planktonic organisms to disperse globally. A global conveyor belt mixes oceanic waters at scales of thousand years (Broecker, 1991). Thus, tectonic and water mass dispersal barriers are often weak and unable to geographically isolate pelagic planktonic populations for extended periods of time (Sexton and Norris, 2008). The geographic distribution of marine planktonic diatoms does not seem to be limited by dispersal; rather it is the environmental selection which dominates diatom community structure (Cermeño and Falkowski, 2009). The general view is that of a broad dispersal of marine planktonic microbes (Cermeño et al., 2010). The picoeukaryotes represent an optimal target to further investigate biogeographical patterns of marine microbes.
In a previous study (Rodríguez-Martínez et al., 2012), we investigated the diversity of an important uncultured picoeukaryote, the MAST-4 (marine stramenopile group 4) lineage (Massana et al., 2004), which is widespread in surface marine waters (except polar systems) and represents approximately 9% of heterotrophic flagellates (Massana et al., 2006, Rodríguez-Martínez et al., 2009). We observed that despite its huge number of cells in the oceans, MAST-4 has a very low genetic divergence and is composed of only five main clades, each representing at least one biological species. The small size (2 μm), high abundance (∼100 cells ml−1 in the ocean surface), worldwide distribution and low genetic diversity make MAST-4 a good example to study marine protist biogeography. In this work, we determined the MAST-4 community structure and distribution by combining automated ribosomal intergenic spacer analysis (ARISA) (Fisher and Triplett, 1999) and 18 S rDNA-ITS1 (Internal Transcribed Spacer 1) gene libraries. MAST-4 diversity was analyzed at 40 different locations, and we found evidence for a strong environmental selection and little dispersal limitation for the most represented clades.
Materials and methods
Study sites and sampling
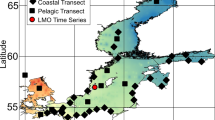
Samples were selected from different oceanographic cruises performed at the North Atlantic Ocean (NOR, NAT, RG, BE and COC), the North Pacific Ocean (WE), the Mediterranean Sea (BL and AL) and the southern hemisphere (IND and DH) (Figure 1). Details of these oligo- and mesotrophic environments (up to 3.4 μg chlorophyll l−1) have been already published: NOR (Not et al., 2005), NAT (González et al., 2000), COC (Alonso-Sáez et al., 2007), WE (del Giorgio et al., 2011), BL (Alonso-Sáez et al., 2008), AL (Arin et al., 2002), IND (Not et al., 2008) and DH (Díez et al., 2004). Seawater was taken with Niskin bottles attached to a CTD (conductivity, temperature, depth recorder) rosette, prefiltered by a 200-μm nylon mesh and then filtered in succession through a 3-μm pore-size polycarbonate filter and a 0.2-μm pore-size Sterivex unit (Durapore, Millipore, Billerica, MA, USA). DNA extraction from the picoplankton (0.2–3 μm) was done using enzymatic and sodium dodecyl sulfate digestion plus phenol purification (Massana et al., 2000). The quality and quantity of extracted genomic DNA was determined with a NanoDrop 1000 (Thermo Fisher Scientific Inc., Wilmington, DE, USA). Physico-chemical data (temperature, salinity) and chlorophyll concentration from the samples were compiled from the previous studies. ARISA fingerprinting was done for the surface samples in all the stations marked in Figure 1, for additional depths in most stations and for a temporal survey at the Blanes Bay Microbial Observatory (BL). Surface samples from stations marked with a star were used for clone libraries.

Global map indicating sampling sites used for ARISA fingerprinting (dots) and sites used for clone libraries (stars). The acronym of the cruise is indicated close to the stations.
Design of PCR primers
Specific primers were designed to amplify the end of the 18S rDNA (290 bp) by the ITS1 and the beginning of the 5.8S (39 bp). The forward primer M4.18S-F (5′-TGGGTAATCTTTGAACGTGAAT-3′), located before the V9 region of the 18S rDNA, was designed based on all MAST-4 sequences for this region available so far (52 unique clones). It had a perfect match (identical in nucleotide sequence) for all these clones, except two (ME1.29 and OLI11066) that had an extra nucleotide (likely a sequencing error). It had >2 mismatches (except for one sequence with one mismatch and three sequences with two mismatches) to non-target sequences from the SILVA database (Pruesse et al., 2007). The reverse primer M4.58S-R (5′-GTTGCGAGAACCTAGAC-3′), located in the 5.8S rDNA, was designed to have a perfect match with the 22 MAST-4 sequences (Rodríguez-Martínez et al., 2012). This primer had at least two mismatches with all stramenopile sequences extracted from GenBank, except for some Labyrinthulida sequences (3 with no mismatches and 62 with one mismatch). Primers were checked for formation of primer dimers, GC (guanine-cytosine) content and theoretical melting temperature in the website www.operon.com, using the Oligo Analysis and Plotting Tool. This primer set gave an amplicon size ranging from 500 to 650 bp.
Construction of clone libraries
The PCR mixture (30 μl) contained 15 ng of DNA template, 0.5 μM of each primer, 200 μM of each dNTP, 1 mM MgCl2, 1.5 units of a Taq DNA polymerase (ThermoPrime, Thermo Scientific, Lafayette, CO, USA) and the enzyme buffer. PCR cycling, carried out in a Bio-Rad thermocycler (Bio-Rad, Hercules, CA, USA), was: initial denaturation at 94 °C for 5 min; 30 cycles with denaturation at 94 °C for 1 min, annealing at 60 °C for 45 s and extension at 72 °C for 1 min; and a final extension at 72 °C for 10 min. We tested the MgCl2 concentration (from 0.5 to 3 mM) and the annealing temperature (from 55 to 66 °C) and chose the most stringent conditions giving the expected band. To check the specificity of the primer set, we confirmed the negative signal with nine non-target cultures (diatoms, haptophytes, dinoflagellates and cyanobacteria). PCR products were purified with the QIAquick PCR Purification kit (QIAGEN, Valencia, CA, USA) and cloned using the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA, USA) with the vector pCR4 following the manufacturer’s recommendations and a vector-insert ratio of 1:5. Putative positive bacterial colonies were picked and transferred to a new LB (Luria-Bertani) plate and finally into LB-glycerol solution for frozen stocks (−80 °C). Presence of correct insert was checked by PCR reamplification with vector primers M13F and M13R using a small aliquot of bacterial culture as template. Amplicons with the right insert size (39–49 clones per library) were sequenced at the Macrogen sequencing service in Korea. Chromatograms were examined with 4Peaks (A. Griekspoor and T. Groothuis, mekentosj.com). Sequences have been deposited in GenBank under accession numbers KC561142–KC561369.
Sequence analysis
Sequences from clone libraries, together with sequences from the ‘SSU-LSU’ dataset (Rodríguez-Martínez et al., 2012), were aligned with MAFFT v6.853 (Katoh and Toh, 2008) with the E-INS-I algorithm, using a MAST-7 sequence as an outgroup. The alignment was inspected visually and modified using secondary structure models folded in mFOLD (Zuker, 2003) as explained before (Rodríguez-Martínez et al., 2012). A maximum likelihood (ML) phylogenetic tree was done with RAxML v7.0.4 MPI version (Stamatakis, 2006), using the General Time Reversible model of nucleotide substitution and a Gamma distributed rate of variation across sites. The shape parameter (α) of the Gamma distribution was estimated from the data set using default options. Phylogenies were done at the University of Oslo Bioportal (www.bioportal.uio.no). One thousand alternative ML trees were run, and the tree with the best likelihood was selected and visualized in FigTree v1.3.1 (Rambaut, 2009). Bootstrap analyses were run with 1000 pseudo-replicates and a consensus tree was constructed with RAxML. To infer intraspecific phylogenies and visualize alternative potential evolutionary paths, we constructed median-joining (MJ) networks (Bandelt et al., 1999) with the Network 4.6.0.0 program (Fluxus Technology, Suffolk, UK). Rarefaction curves for each clone library were constructed using Mothur (Schloss et al., 2009). The genetic differentiation between populations was estimated with DNAsp 5.10.1 using the fixation index (Fst), which ranges from 0 (no genetic differentiation) to 1 (complete differentiation).
Generation of ARISA profiles
Environmental DNA samples were PCR-amplified in triplicate for ARISA in a MJ Research cycler. PCR conditions were the same as described before except that we used a volume of 25 μl with 10 ng of DNA template, a different Taq DNA Polymerase (‘Gene Choice’) and the forward primer was fluorescently labeled (5-HEX). PCR products stored in the dark at 4 °C were purified with MultiScreen PCRμ96 Plates and quantified using PicoGreen fluorescence (Invitrogen) in a SpectraMax M2 microplate reader (Molecular Devices Corp., Sunnyvale, CA, USA). Ten ng DNA were ethanol precipitated from triplicates or from pooled PCR products (when the yield of the PCR was low), followed by resuspension with 0.078 μl Tween, 9.67 μl water and 0.25 μl fluorescently labeled internal size standard, CST ROX 60-1500 bp (http://www.bioventures.com/). Samples were run on a MegaBACE 1000 automated capillary sequencer (Molecular Dynamics, Sunnyvale, CA, USA). The electropherograms were then analyzed using DAx software (v8.0; Van Mierlo Software Consultancy, Eindhoven, the Netherlands). Only peaks exceeding four times the noise signal of the electropherogram curve were considered.
Analysis of fingerprinting data
From DAx output tables, peak heights were binned using the ‘fixed window’ binning strategy to take into account the size-calling imprecision from ARISA fingerprints (Hewson and Fuhrman, 2006). In order to determine the best window size with our data, we applied the ‘automatic binning algorithm’ (Ramette, 2009) developed in a R script (The R Foundation for Statistical Computing (http://cran.r-project.org/)); we chose 2 bp. To identify the best window frame (out of the 20 possible starting with a shift value of 0.1), we used the ‘interactive binning algorithm’ (Ramette, 2009). This algorithm binned the peaks for each frame, calculated the relative fluorescence intensity of each binned peak by dividing its height by the total peak height of the sample and omitted peaks with values <0.5% (considered as background). We added an option in the script to compare frames considering only triplicate samples (instead of all the samples). The frame with the best correlation among triplicates was chosen; starting with 1.3 in our case. The final output was a table with the relative intensity of each binned peak (each considered as a different operational taxonomic unit (OTU)) in the size range of 500–650 bp. We then performed a permutational multivariate analysis of variance (PERMANOVA) test using the sample as a grouping factor in order to estimate the variability due to the experimental error. If triplicates were identical, this test would explain 100% of the variability. In our case, it explained 92%, indicating that only 8% of the variability of the samples was due to technical imprecision.
A consensus OTU-table (relative intensity of all the OTUs in all the samples) was obtained by averaging the triplicates in each sample, as long as OTUs appeared at least in two of the three replicates. A distance matrix from the consensus OTU-table was calculated with the Bray–Curtis dissimilarity index (community distance matrix). Patterns were explored using clustering analysis (along with the SIMPROF (similarity profile) significance test) and nonmetric multidimensional scaling (NMDS) analysis. A stress value was calculated based on the differences between Bray–Curtis distances and actual distances on the MDS plot, with lower values depicting a better representation of sample distances in a two-dimensional space. Simple and partial Mantel tests (with the Pearson correlation and based on 999 permutations) were done to compare the community distance matrix with a geographical distance matrix (using geographical coordinates in a perfect sphere) and a temperature Euclidean distance matrix. The relationship between community composition (the OTU-table) and environmental factors was analyzed by a Constrained Correspondence Analysis. Automatic forward selection with significance tests of Monte Carlo permutations were used to build the optimal models. Additionally, we did a PERMANOVA test to check the importance of the environmental factors in the community composition. We also assessed the contribution of each OTU to the observed similarity (or dissimilarity) between groups with a similarity percentage (SIMPER) analysis. All multivariate analyses were also done with an OTU-table, including triplicates and with transformed data (arcsine of the square root of the relative intensity) to reduce the skew; no differences in the results were seen (data not shown). Statistical tests and graphics were done using R packages (Gmt, Vegan, MASS), CANOCO 4.5 for Windows (ter Braak and Smilauer, 2002) and Primer v6.1.2 software.
Results
Validation of the different MAST-4 clades
In a previous study, we showed that MAST-4 was composed of five main clades (Rodríguez-Martínez et al., 2012). To verify the robustness of these clades, we obtained 228 additional sequences of 500–650 bp encompassing the V9 region of the 18S rDNA and the ITS1 region from five different locations (Figure 1), including the locations from the previous study and one additional site. All sequences obtained in clone libraries belonged to the MAST-4 lineage, demonstrating the specificity of the primers. The ML phylogenetic tree (Figure 2) revealed that the new sequences were distributed in the five clades previously observed, sometimes forming new subclades. Because we used the very variable ITS1 region, bootstrap values for the deepest branches were low, but the purpose behind this locus choice was the definitive assignment of sequences to clades and subclades, rather than a robust interclade topology. Sequences grouped in 12 different helix III types for the ITS1 secondary structure as described in Rodríguez-Martínez et al. (2012, Figure 2 and Supplementary Figure S1). Clade A included sequences from the five different sites, all with the same helix III motif. Clade B also had one helix III motif but was divided in three phylogenetic subclades. Clade C presented four helix III motifs, including several CBCs (compensatory base changes) that corresponded well with phylogenetic subclades except for subclade C2 that had two motifs differing by a hemiCBC. Subclades C2 and C3 included sequences from only one location. Clade D had only four sequences but represented two helix III motifs. Finally, clade E consisted of four helix III motifs, two of them forming well-differentiated lineages (E2 and E3), and the other two, with only one hemiCBC in the fifth base pair, included in the E1 subclade. Rarefaction curves of the five libraries did not reach a clear saturation at a level of sequence similarity of 99%, indicating undersampling of the community (data not shown).

Maximum likelihood phylogenetic tree of MAST-4 built with 228 new sequences (end of 18S rDNA and complete ITS1) and 22 clones from a previous study (indicated with letters). Clades and subclades are indicated with gray areas. Bootstraps values >50% are shown. The scale bar indicates 0.2 substitutions per position. MJ networks appear at the right of the tree, with ribotypes in a different color depending the sample. Scale bars indicate 10 base changes between ribotypes (note different scale for each clade). At the top right, sequences for the conserved helix III stem (derived from ITS1 secondary structures) are listed.
Biogeographical patterns from sequence analyses
To examine biogeographical patterns from the five samples, we used the ITS1 sequences to construct MJ networks for each clade (Figure 2, right panels), which allow visualization of potential evolutionary paths in data sets with large sample sizes and small genetic distances between individuals. These networks revealed variation in genetic diversity in each clade. According to this analysis, sequences within clades E and A were very similar, whereas clade B exhibited a larger diversity. In a second step, we used the ITS1 libraries to calculate Fst values a measure of the genetic differentiation among populations from different sites. This was done for each clade separately, as they form separate putative species.
Clade A included sequences from all the five sites, and there were few base changes between ribotypes as evidenced by short connecting lines in the MJ network. Fst values were generally low between the different geographical populations of clade A (Figure 3). The Indian population (IND) appeared the most isolated, with Fst values around 0.5 with other locations, whereas the other four populations had lower pairwise Fst values (<0.22). Interestingly, the two populations in the warm sites (Sargasso Sea (BE) and Mediterranean (BL)) showed very little differentiation, as occurred between the two populations from the cold sites (North Pacific (WE) and North Atlantic (NOR)). In contrast to clade A, the MJ network for clades B and C showed a strong spatial structuring, with sequences from the different locations typically forming separate subclades (Fst values >0.80 indicated well-differentiated populations in each site). Subclade C1 stands as one exception for this trend, as little differentiation was seen between BE and IND populations (Fst=0.18). Finally, sequences from subclade E1, originating from three different regions, were well mixed in the network plot, and there were two cases of identical sequences from distant sites (Figure 2). The Fst values between these populations were low, particularly between WE and NOR sites (Figure 3).

Population differentiation of clades A and E1 among the five sites with clone libraries. Fst values for clades A (first value) and E1 (second value, when available) are shown above the lines, with line thickness indicating the similarity in population composition (low Fst values).
Agreement between ARISA profiles and clone libraries
In order to interpret ARISA data, we first analyzed the fingerprints from the same samples that originated the clone libraries. For each sample, ARISA profiles were similar to the size distribution of clones in the corresponding library (Supplementary Figure S2), suggesting that all peaks derived from MAST-4 phylotypes (although ARISA peaks were estimated at 5–9 bp larger than the actual fragment size). We then combined all clones from the different libraries to obtain a taxonomic assignment of ARISA peaks (Figure 4). Clones from clades A and C overlapped at the 511–537 bp region, although some sizes were exclusive to a given clade, such as 513, 525 and 528 (clade A) or 517 and 519 (clade C). Clones from clade B exhibited a wider size range, from 527 to 590 bp, and were the only ones occupying the size spectra from 560 to 574 bp. Clade D, with only four sequences, had the largest clones (648 bp) and also appeared in the 580–594 region intermixed with other clades. Clade E had clones in a restricted region (580–584 bp), and the majority were 581 bp. This analysis enabled us to identify or partially identify clades in subsequent ARISA profiles.

Summary of clone sizes for the five libraries (228 sequences) colored/patterned according to the corresponding MAST-4 clade.
Biogeographical patterns from ARISA fingerprints
We investigated the biogeographical patterns of MAST-4 assemblages by comparing the ARISA fingerprints from 107 samples obtained from 40 separated sites. For most sites (25 out of 40), there were several depths sampled (subsurface to 100 or 250 m) and a subset of 23 samples derived from a temporal study in the Mediterranean site (BL). Samples were analyzed in triplicate and then averaged for further statistical analyses as they were highly similar (see Materials and methods). The correlation between community and geographical distance matrices done with a Mantel test (Figure 5a) was weak and significant (r=0.28, P=0.001), suggesting that geographical distance did not have a strong relation with sample composition. A partial Mantel test with geographical distance conditioned by temperature showed a weaker correlation (r=0.14, P=0.001) giving even less weight to the relationship between community and geographical distance, once the effect of the main environmental variable (see below) was removed. Interestingly, a group of samples from distant sites (∼15 000 km) were highly similar, whereas samples from the same geographical location could be both similar or different. A Mantel test comparing the community and temperature distances (Figure 5b) showed a high and significant correlation (r=0.60, P=0.001), highlighting temperature as an important driver of community composition. Moreover, a partial Mantel test with temperature distance conditioned by geographical distance did not bring down the correlation (r=0.57, P=0.001). Analysis of community composition by NMDS revealed a clear grouping of samples based on temperature (Figure 5c) and no trend for geographical location (Supplementary Figure S3). To create these groups, we used a separate dendrogram (not shown) where cold samples (from 2 to 9.4 °C) grouped in a cluster with 82% similarity, and the temperate (from 9.5–16.9 °C) and warm (from 17–30 °C) groups also appeared, although with some exceptions. The boundary between the temperate and warm groups was obtained from the clustering of BL samples (see below).

Analysis of the global data set of ARISA fingerprints for MAST-4 diversity. (a) Plot comparing community distances (calculated from the OTU-table) and geographic distances among samples, together with the Mantel test statistic r. (b) Plot comparing community and environmental (temperature) distances, together with the Mantel test statistic r. (c) NMDS diagram displaying each sample as a function of their community distances. Different symbols represent the temperature grouping. (d) Constrained Correspondence Analysis diagram displaying each sample in function of their community distances and constrained for the three most important factors: temperature (Temp), salinity (Sal) and sampling depth (Z). Arrows represent the direction and magnitude of the environmental factor gradient.
We performed several analyses to identify and quantify the factors driving community changes. A Constrained Correspondence Analysis (Figure 5d; Table 1) confirmed that temperature was the factor explaining most of the variance (51%), whereas sampling depth (Z) explained 18% of the variance. The six deepest samples (200 and 250 m) collected from the Indian Ocean and the Mediterranean Sea are grouped together in the top-left of the plot. These variables were followed by salinity, bottom depth (Zmax, a proxy for coastal vs offshore waters) and chlorophyll concentration. The sum of these factors explained 91% of the variability. When samples within each temperature group were analyzed separately, temperature was less important than salinity and sampling depth. A second analysis was done after grouping the factors at discrete levels. A PERMANOVA test considering grouped factors showed that temperature explained 36% of the variability in community composition, whereas all factors together explained 55% (Table 1).
We then searched for the OTUs driving the differences among cold, temperate and warm samples. The SIMPER test identified five OTUs with a dissimilarity contribution between groups >5% (Table 2). OTU 589 had the largest dissimilarity contribution in all pairwise analyses, especially when including cold samples. This was consistent with the 99% similarity contribution of OTU 589 to the cold group (Table 2) and its predominant presence in cold waters (Figure 6). OTU 527 was the most important in warm samples (22% similarity contribution) and contributed 7% to the dissimilarity between all comparisons with warm samples. OTUs 529 and 531 appeared important in temperate and warm samples, whereas OTU 567 was characteristic for temperate samples and absent in cold samples. Using the agreement between ARISA and clone libraries peak profiles, we predicted that OTU 589 corresponded to a clone of 581 bp in size, belonging to clade E, whereas OTUs 527, 529 and 531 (clone sizes between 519 and 524 bp) could belong to either clades A or C and OTU 567 (clone size 560 bp) belonged to clade B. Therefore, clade E was best adapted to cold water, clades A and/or C were typical of temperate and warm waters while clade B was most characteristic of temperate waters.

Relative intensity of the most important OTUs in the ARISA fingerprints in all the samples displayed according to temperature. Gray circles represent samples where the OTU was not detected.
Temporal changes at Blanes Bay
At the Blanes Bay (BL) station in the NW Mediterranean Coast, we analyzed 23 different ARISA fingerprints covering a monthly seasonal sampling in 2003 and random dates from 2001 to 2006. Samples exhibited a large variability in the composition of MAST-4 and grouped, in an associated dendrogram (not shown), in two clusters differentiated by seawater temperature (> or <17 °C), as highlighted in the NMDS plot (Figure 7). The similarity of temperate samples was >58% and the similarity of warm samples was > 37%. Each group contained samples from different years, highlighting the importance of temperature in defining community composition along this temporal scale.

NMDS analysis of ARISA fingerprints in the temporal study at Blanes Bay. Samples are displayed with symbols for temperature as in Figure 5 and with the date of sampling (month–year). Points enclosed by dashed and solid lines cluster at 37% and 58% similarity, respectively, in a separate dendrogram (not shown).
The PERMANOVA test for this subset of samples, considering the same groups as above, showed that temperature was also the factor explaining most of the community composition variability (46%), a contribution even higher than when considering all samples. The SIMPER test revealed that OTU 589 (clade E), which was predominant in temperate waters, had again the largest dissimilarity contribution (30%) between temperate and warm samples. Moreover, this analysis highlighted additional OTUs responsible for community changes, such as OTU 593 that was characteristic of warm samples and contributed 8% of the dissimilarity between the temperature groups.
Discussion
Despite the recent interest in microbial biogeography, the field has suffered from conceptual and methodological limitations that confuse the emerging conclusions (Martiny et al., 2006). The first problem is the analytical strategy. Many studies are based on a handful of isolated strains from separate geographical sites (Ki and Han, 2005; Kooistra et al., 2008), which can skew the conclusions reached about biogeography. Here rather than relying on culturing, we employ molecular markers easily amplified from environmental DNA. Thus the sampled diversity is more representative of the natural assemblage (by avoiding culturing bias). In addition, many more individuals (sequences) can be retrieved at once, allowing for more robust population comparisons.
Second, it is very important to choose a good taxonomic marker, because the view that no biogeographical patterns exist in microorganisms (Finlay et al., 2006) could be caused by an ambiguous or incorrect identification of protist species (Lomolino et al., 2006). Indeed many protist species appear to be widely distributed when identified via morphology, but these ‘morphological species’ most likely include cryptic species (Dolan, 2006). In our study, we used the highly variable ITS1 region as a diversity marker, which provides an enhanced taxonomic resolution as compared with the 18S rDNA gene (Brown and Fuhrman, 2005). ITS1 has been used to assess the inter- and intraspecific diversity of Pseudo-nitzschia populations (Orsini et al., 2004) and for elucidating Pseudo-nitzschia community structure using ARISA fingerprints (Hubbard et al., 2008). Our analysis using both the ITS1 sequences and ARISA fingerprints confirmed the ITS region as a good marker for studying microbial biogeography.
Third, it is important to clearly determine the genetic diversity of the studied taxa. We focus here on the uncultured protist MAST-4, which exhibits a limited evolutionary diversification (Rodríguez-Martínez et al., 2012). By designing specific primers amplifying the ITS1 region of MAST-4, we increased the currently available sequences for this group by 10-fold. Even with this order of magnitude increase in sequence sampling, only two additional putative species were revealed within this group (based on the conserved regions of ITS secondary structures (Coleman, 2009)). The evidence for not more than 12 putative species confirms the low diversity of MAST-4. One of the clades, clade A, still appears to be a single species, with almost all clones having the same helix III motif in the ITS1 secondary structure. This was consistent with our previous study showing low-sequence divergence, incongruent tree topologies and no polymorphisms in the critical regions of the ITS1 and ITS2 secondary structures for sequences within this clade (Rodríguez-Martínez et al., 2012). With the same criteria, subclade E1 also forms a single species. The structuring of MAST-4 in a few genetically distinct species is similar to that found in other marine protists, such as the cosmopolitan diatom Skeletonema costatum (Kooistra et al., 2008).
Finally, a limitation of many studies is the inability to disentangle the relative contribution of contemporary environmental conditions and historical contingencies in shaping the spatial variations of microbial communities (Martiny et al., 2006). Our study addresses this question by analyzing spatial changes together with the associated environmental factors. In addition, as a single sampling event may not cover the complete biodiversity of a given location (Nolte et al., 2010), we also evaluated temporal changes over an annual cycle.
The analysis of MAST-4 assemblages using ARISA fingerprints showed that temperature was the main factor influencing the distribution patterns, as has been observed in other marine microbes like Prochlorococcus (Martiny et al., 2009) and bacterial and archaeal assemblages (Winter et al., 2008). Distant samples sharing the same temperature could have similar MAST-4 composition, whereas samples with different temperatures contained very different MAST-4 assemblages. This global pattern was also observable in a temporal study at the same geographical site (Blanes Bay). In this location with a thermal seasonal cycle from 12 to 24 °C, samples again grouped by temperature, confirming it as an important structuring factor. Moreover, the Fst analysis revealed very little differentiation between populations geographically distant but with a similar temperature. It is worth mentioning that a similar pattern could have been obtained due to another driving factor not measured here but strongly correlated with temperature (Martiny et al., 2009). Thus, the spatial distribution of MAST-4 seemed to be mainly controlled by contemporary environmental factors with a null or low degree of provincialism. Other abiotic factors that are known to be important in defining microbial community composition, such as salinity (Logares et al., 2009) and sampling depth (Winter et al., 2008), were important within each of the defined temperature group.
In a more detailed analysis of clades, those which likely constitute a single species (A and E1) displayed well-mixed populations in the MJ networks and exhibited low differentiation (Fst values) among populations, particularly in samples with similar temperature regardless of whether they came from distant places. By contrast, clades B and C, each probably composed of more than one species, exhibited some spatial structuring, with subclades appearing in only one library. This intriguing feature, perhaps indicating some dispersal restriction for clades B and C, could also be due to undersampling and deserves more attention in future surveys. If confirmed by further results, the dispersal limitation of clades B and C would be comparable with that found with the cosmopolitan marine planktonic diatom Pseudo-nitzschia pungens (Casteleyn et al., 2010).
Our data also highlighted specific MAST-4 lineages adapted to different temperature regimes. Thus, clade B, and either or possibly both clades A or C, were characteristic of temperate and warm waters, whereas clade E1 (represented by OTU 589) was adapted to inhabit cold waters. The genetic structure of MAST-4 with different lineages, some ubiquitous in the oceans and with particular ecological properties, resembles that of other marine microorganisms. Thus, different Ostreococcus (Rodríguez et al., 2005) and Synechococcus lineages (Ahlgren and Rocap, 2006) are adapted to different light levels, and a Micromonas pusilla clade adapted to cold temperature has been reported (Lovejoy et al., 2006). It has been proposed that this ecotypic differentiation can partly explain the success of these microorganisms, allowing them to exploit the whole spectrum of habitat variability.
The uncultured free-living protist MAST-4 is widely abundant and very small and thus possesses the properties for a worldwide distribution (Finlay, 2002). Moreover, it lives in a marine habitat, where wind, waves and currents produce mixing events that facilitate the dispersion. However, we did not observe all MAST-4 clades at all locations but saw biogeographical patterns, stressing the importance of the end of the tenet, ‘but the environment selects’. It is reasonable to hypothesize that MAST-4 has a huge dispersal capacity and can arrive everywhere within a marine habitat. For instance, there is one record of a MAST-4 sequence in the Arctic Ocean (Lovejoy and Potvin, 2011), showing the potential to arrive to such high latitudes probably dragged by Pacific coastal currents. But then, depending on the environmental conditions, different organisms will thrive, resulting in different community patterns. Although microorganisms could spread across all suitable habitats, local adaptations eventually may promote speciation (Medlin, 2007). In conclusion, we did not see strong marine geographical barriers for the dispersal of MAST-4, instead temperature appeared as the main driver for community composition. For MAST-4, we suggest that the environment makes a taxonomic selection among clades with distinct physiological adaptations.
Accession codes
References
Ahlgren NA, Rocap G . (2006). Culture isolation and culture-independent clone libraries reveal new marine Synechococcus ecotypes with distinctive light and N physiologies. Appl Environ Microbiol 72: 7193–7204.
Alonso-Sáez L, Arístegui J, Pinhassi J, Gómez-Consarnau L, González JM, Vaqué D et al (2007). Bacterial assemblage structure and carbon metabolism along a productivity gradient in the NE Atlantic Ocean. Aquat Microb Ecol 46: 43.
Alonso-Sáez L, Vázquez-Domínguez E, Cardelús C, Pinhassi J, Sala MM, Lekunberri I et al (2008). Factors controlling the year-round variability in carbon flux through bacteria in a coastal marine system. Ecosystems 11: 397–409.
Arin L, Moran X, Estrada M . (2002). Phytoplankton size distribution and growth rates in the Alboran Sea (SW Mediterranean): short term variability related to mesoscale hydrodynamics. J Plankton Res 24: 1019.
Baas-Becking LGM . (1934). Geobiologie of inleiding tot de milieukunde. WP Van Stoc- kum & Zoon (in Dutch): The Hague, the Netherlands 18.
Bandelt H, Forster P, Röhl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Beijerinck MW . (1913). De infusies en de ontdekking der backteriën. Jaarboek van de Koninklijke Akademie van Wetenschappen. Müller: Amsterdam, The Netherlands, pp 1–28.
Broecker WS . (1991). The great ocean conveyor. Oceanography 4: 79–89.
Brown M, Fuhrman J . (2005). Marine bacterial microdiversity as revealed by internal transcribed spacer analysis. Aquat Microb Ecol 41: 15–23.
Casteleyn G, Leliaert F, Backeljau T, Debeer A, Kotaki Y, Rhodes L et al (2010). Limits to gene flow in a cosmopolitan marine planktonic diatom. Proc Natl Acad Sci USA 107: 12952–12957.
Cermeño P, Falkowski PG . (2009). Controls on diatom biogeography in the ocean. Science 325: 1539–1541.
Cermeño P, De Vargas C, Abrantes F, Falkowski PG . (2010). Phytoplankton biogeography and community stability in the ocean. PLoS ONE 5: e10037.
Coleman AW . (2009). Is there a molecular key to the level of “biological species” in eukaryotes? A DNA guide. Mol Phylogenet Evol 50: 197–203.
del Giorgio PA, Condon R, Bouvier T, Longnecker K, Bouvier C, Sherr E et al (2011). Coherent patterns in bacterial growth, growth efficiency, and leucine metabolism along a northeastern Pacific inshore–offshore transect. Limnol Oceanogr 56: 1–16.
Díez B, Massana R, Estrada M, Pedrós-Alió C . (2004). Distribution of eukaryotic picoplankton assemblages across hydrographic fronts in the Southern Ocean, studied by denaturing gradient gel electrophoresis. Limnol Oceanogr 49: 1022–1034.
Dolan J . (2006). Microbial biogeography? J Biogeogr 33: 199–200.
Fenchel T, Finlay BJ . (2004). The ubiquity of small species: patterns of local and global diversity. Bioscience 54: 777–784.
Finlay BJ . (2002). Global dispersal of free-living microbial eukaryote species. Science 296: 1061–1063.
Finlay BJ, Esteban G, Brown S, Fenchel T, Hoef-Emden K . (2006). Multiple cosmopolitan ecotypes within a microbial eukaryote morphospecies. Protist 157: 377–390.
Fisher MM, Triplett EW . (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol 65: 4630–4636.
González JM, Simó R, Massana R, Covert JS, Casamayor EO, Pedrós-Alió C et al (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol 66: 4237–4246.
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M et al (2004). Spatial scaling of microbial eukaryote diversity. Nature 432: 747–750.
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH . (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10: 497–506.
Hewson I, Fuhrman J . (2006). Improved strategy for comparing microbial assemblage fingerprints. Microb Ecol 51: 147–153.
Horner-Devine MC, Silver JM, Leibold MA, Bohannan BJM, Colwell RK, Fuhrman JA et al (2007). A comparison of taxon co-occurrence patterns for macro- and microorganisms. Ecology 88: 1345–1353.
Hubbard K, Rocap G, Armbrust E . (2008). Inter- and intraspecific community structure within the diatom genus Pseudo-nitzschia (Bacillariophyceae). J Phycol 44: 637–649.
Katoh K, Toh H . (2008). Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9: 286–298.
Ki J, Han M . (2005). Molecular analysis of complete SSU to LSU rDNA sequence in the harmful dinoflagellate Alexandrium tamarense (korean isolate, HY970328M). Ocean Sci J 40: 43–54.
Kooistra WH, Sarno D, Balzano S, Gu H, Andersen RA, Zingone A . (2008). Global diversity and biogeography of Skeletonema species (Bacillariophyta). Protist 159: 177–193.
Logares R, Bråte J, Bertilsson S, Clasen JL, Shalchian-Tabrizi K, Rengefors K . (2009). Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol 17: 414–422.
Lomolino MV, Riddle BR, Brown JH . (2006) Biogeography, 3rd edn, Sinauer Associates, Inc.: Sunderland, MA, USA.
Lovejoy C, Massana R, Pedrós-Alió C . (2006). Diversity and distribution of marine microbial eukaryotes in the Arctic Ocean and adjacent seas. Appl Environ Microbiol 72: 3085–3095.
Lovejoy C, Potvin M . (2011). Microbial eukaryotic distribution in a dynamic Beaufort Sea and the Arctic Ocean. J Plankton Res 3: 431–444.
MacArthur RH, Wilson EO . (1967) The Theory of Island Biogeography. Princeton University Press: Princeton, NJ, USA.
Martiny AC, Tai APK, Veneziano D, Primeau F, Chisholm SW . (2009). Taxonomic resolution, ecotypes and the biogeography of Prochlorococcus. Environ Microbiol 11: 823–832.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Massana R, Castresana J, Balagué V, Guillou L, Romari K, Groisillier A et al (2004). Phylogenetic and ecological analysis of novel marine stramenopiles. Appl Environ Microbiol 70: 3528–3534.
Massana R, DeLong EF, Pedrós-Alió C . (2000). A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl Environ Microbiol 66: 1777–1787.
Massana R, Terrado R, Forn I, Lovejoy C, Pedrós-Alió C . (2006). Distribution and abundance of uncultured heterotrophic flagellates in the world oceans. Environ Microbiol 8: 1515–1522.
Medlin L . (2007). If everything is everywhere, do they share a common gene pool? Gene 406: 180–183.
Nolte V, Pandey RV, Jost S, Medinger R, Ottenwälder B, Boenigk J et al (2010). Contrasting seasonal niche separation between rare and abundant taxa conceals the extent of protist diversity. Mol Ecol 19: 2908–2915.
Not F, Latasa M, Scharek R, Viprey M, Karleskind P, Balagué V et al (2008). Protistan assemblages across the Indian Ocean, with a specific emphasis on the picoeukaryotes. Deep-Sea Res I 55: 1456–1473.
Not F, Massana R, Latasa M, Marie D, Colson C, Eikrem W et al (2005). Late summer community composition and abundance of photosynthetic picoeukaryotes inNorwegian and Barents Seas. Limnol Oceanogr 50: 1677–1686.
O’Malley MA . (2008). ‘Everything is everywhere: but the environment selects': ubiquitous distribution and ecological determinism in microbial biogeography. Stud His Phil Biol Biomed Sci 39: 314–325.
Orsini L, Procaccini G, Sarno D, Montresor M . (2004). Multiple rDNA ITS-types within the diatom Pseudo-nitzschia delicatissima (Bacillariophyceae) and their relative abundances across a spring bloom in the Gulf of Naples. Mar Ecol Prog Ser 271: 87–98.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Rambaut A . (2009) FigTree. Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, http://tree.bio.ed.ac.uk/software/figtree.
Ramette A . (2009). Quantitative community fingerprinting methods for estimating the abundance of operational taxonomic units in natural microbial communities. Appl Environ Microbiol 75: 2495–2505.
Rodríguez F, Derelle E, Guillou L, Le Gall F, Vaulot D, Moreau H . (2005). Ecotype diversity in the marine picoeukaryote Ostreococcus (Chlorophyta, Prasinophyceae). Environ Microbiol 7: 853–859.
Rodríguez-Martínez R, Labrenz M, Del Campo J, Forn I, Jürgens K, Massana R . (2009). Distribution of the uncultured protist MAST-4 in the Indian Ocean, Drake Passage and Mediterranean Sea assessed by real-time quantitative PCR. Environ Microbiol 11: 397–408.
Rodríguez-Martínez R, Rocap G, Logares R, Romac S, Massana R . (2012). Low evolutionary diversification in a widespread and abundant uncultured protist MAST-4). Mol Biol Evol 29: 1393–1406.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Sexton PF, Norris RD . (2008). Dispersal and biogeography of marine plankton: long-distance dispersal of the foraminifer Truncorotalia truncatulinoides. Geology 36: 899–902.
Stamatakis A . (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
ter Braak C, Smilauer P . (2002) CANOCO Reference Manual on CanoDraw for Windows User’s Guide: Software for Canonical Community Ordination (version 4.5). Microcomputer Power: Ithaca, NY, USA.
Vyverman W, Verleyen E, Sabbe K, Vanhoutte K, Sterken M, Hodgson DA et al (2007). Historical processes constrain patterns in global diatom diversity. Ecology 88: 1924–1931.
Whitaker RJ . (2003). Geographic barriers isolate endemic populations of hyperthermophilic Archaea. Science 301: 976–978.
Whitfield J . (2005). Biogeography: is everything everywhere? Science 310: 960–961.
Winter C, Moeseneder MM, Herndl GJ, Weinbauer MG . (2008). Relationship of geographic distance, depth, temperature, and viruses with prokaryotic communities in the Eastern Tropical Atlantic Ocean. Microb Ecol 56: 383–389.
Zuker M . (2003). mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406.
Acknowledgements
Funding has been provided by FPI fellowships from the Spanish Ministry of Education and Science to RRM and GS and by projects FLAME (CGL2010-16304, MICINN, Spain) to RM, and ATOL (NSF, 0629521) to GR. We thank R.E. Collins for DAx software help, E. Sañé for Primer software help, R. Logares for statistical advices and C. Williams for molecular help.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Rodríguez-Martínez, R., Rocap, G., Salazar, G. et al. Biogeography of the uncultured marine picoeukaryote MAST-4: temperature-driven distribution patterns. ISME J 7, 1531–1543 (2013). https://doi.org/10.1038/ismej.2013.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.53
Keywords
This article is cited by
-
How Communities of Marine Stramenopiles Varied with Environmental and Biological Variables in the Subtropical Northwestern Pacific Ocean
Microbial Ecology (2022)
-
Environmental and Spatial Influences on Biogeography and Community Structure of Saltmarsh Benthic Diatoms
Estuaries and Coasts (2021)
-
Disentangling the mechanisms shaping the surface ocean microbiota
Microbiome (2020)
-
Accessing the genomic information of unculturable oceanic picoeukaryotes by combining multiple single cells
Scientific Reports (2017)
-
Community patterns and temporal variation of picoeukaryotes in response to changes in the Yellow Sea Warm Current
Journal of Oceanography (2017)
