
Abstract
Microbial communities have a key role in the physiology of the sponge host, and it is therefore essential to understand the stability and specificity of sponge–symbiont associations. Host-specific bacterial associations spanning large geographic distance are widely acknowledged in sponges. However, the full spectrum of specificity remains unclear. In particular, it is not known whether closely related sponges host similar or very different microbiota over wide bathymetric and geographic gradients, and whether specific associations extend to the rare members of the sponge microbiome. Using the ultra-deep Illumina sequencing technology, we conducted a comparison of sponge bacterial communities in seven closely related Hexadella species with a well-resolved host phylogeny, as well as of a distantly related sponge Mycale. These samples spanned unprecedentedly large bathymetric (15–960 m) gradients and varying European locations. In addition, this study included a bacterial community analysis of the local background seawater for both Mycale and the widespread deep-sea taxa Hexadella cf. dedritifera. We observed a striking diversity of microbes associated with the sponges, spanning 47 bacterial phyla. The data did not reveal any Hexadella microbiota co-speciation pattern, but confirmed sponge-specific and species-specific host–bacteria associations, even within extremely low abundant taxa. Oligotyping analysis also revealed differential enrichment preferences of closely related Nitrospira members in closely related sponges species. Overall, these results demonstrate highly diverse, remarkably specific and stable sponge–bacteria associations that extend to members of the rare biosphere at a very fine phylogenetic scale, over significant geographic and bathymetric gradients.
Similar content being viewed by others

Introduction
Interactions between symbiotic microbes and the hosts they colonize influence host health, reproduction and nutrition (Gootenberg and Turnbaugh, 2011; Proctor, 2011; McFall-Ngai et al., 2013). The sponge holobiont (that is, the hosts and their symbionts as functional entities) represents one of the oldest symbioses between microbes and metazoans and offers a unique model to study marine animal symbioses. Microbial communities appear to be essential for the fitness of the sponge host. Sponge-associated microbial communities affect diverse host metabolic functions including nitrogen, carbon and sulfur cycling. They can provide additional nutrients and produce secondary metabolites that improve the defense of the host against predation, fouling and diseases (Hentschel et al., 2006; Taylor et al., 2007a). Several recent studies also correlate environmental parameters such as elevated seawater temperatures (Webster et al., 2008; Cebrian et al., 2011) or the presence of heavy metals (Webster et al., 2001; Selvin et al., 2009) with shifts in microbial communities and declines in sponge health.
The advent of DNA sequencing methods has enabled important advances in sponge microbiome studies. Deep sequencing with next-generation platforms has revealed bacterial diversity that is unparalleled in any invertebrate host and approaches the complexity of the human gut (Webster et al., 2010). In line with the previously described ‘sponge-specific’ clusters (Hentschel et al., 2002; Taylor et al., 2007a), studies using next-generation sequencing have shown sponge-specific bacterial communities in a number of sponge species (Lee et al., 2011; Schmitt et al., 2012). In addition, the observation of specific microbial communities at finer host phylogenetic levels, for example, within each sponge species (Lee et al., 2011), suggests potential host-symbiont co-speciation.
Using the molecular fingerprinting approach ARISA (Automated Ribosomal Intergenic Spacer Analysis), Schöttner et al., (2013) observed a significant relationship between bacterial community dissimilarity and host phylogenetic distance. This study contrasted with the lack of co-speciation highlighted by Schmitt et al. (2012), who employed 16S rDNA pyrosequencing analyses. These discrepancies may reflect the choice and length of different molecular markers, and neither study used deep sequencing of host-microbiota with in-depth phylogeny of host sponge species. Deep sequencing has also detected sponge-associated microbial organisms in the surrounding water column, but at very low abundances (Webster et al., 2010; Taylor et al., 2012). This rare seawater biosphere might serve as a seed bank for colonization of sponges, implying that environmental transmission may have a significant role in the acquisition of symbionts by sponges. These studies indicate that complex ecological and evolutionary factors influence the stability and specificity of sponge–symbiont associations.
The goal of this work was to explore host-specificity patterns over uniquely wide bathymetric gradients and varying geographic locations, in both dominant and rare bacterial members of the sponge microbiome. The sponge-associated bacterial communities in seven congeneric (and cryptic) encrusting Hexadella (Porifera: Verongida) sponge species from shallow and deep-sea European coral environments (mainly built by the species Lophelia pertusa (Linnaeus 1758) and Madrepora oculata, Linnaeus 1758) were examined in conjunction with a fully resolved phylogeny of the host organisms. These bacterial communities were also compared with the microbiomes of the distantly related sponge Mycale sp. that occur sympatrically with Hexadella cf. dedritifera. Finally, we characterized microbial communities from local ambient seawater to further assess the influence of the environment (that is, biotic component) versus the sponge host on the composition and structure of the associated bacterial community. Ultra-deep sequencing permitted the detection of very low-abundance organisms that make up the rare sponge biosphere, and the elucidation of host-specificity patterns of dominant and functionally important members of the sponge microbiota.
Materials and methods
Sample collection, DNA extraction and cytochrome c oxidase subunit I amplification
The Hexadella species were collected at various locations from 15 to 960 m (Table 1 and Figure 1) on the European margins, from the Mediterranean to the Greenland Sea and were preserved in absolute ethanol and stored at room temperature before DNA extraction. Their phylogenetic relationships inferred from comparisons of mitochondrial and nuclear genes were previously reported (Reveillaud et al., 2010). The taxonomy for shallow-water species Hexadella pruvoti, Hexadella crypta, Hexadella racovitzai and Hexadella topsenti were also previously analyzed using molecular data, morphology, toxicity and biochemistry (Reveillaud et al., 2012).

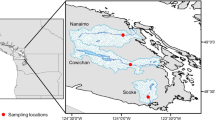
Map displaying sampling locations of deep-sea sites (star shape) and shallow-water ones (square shape). The boundary between shallow-water and deep sea was defined at a depth of 50 m, as previously used for corals (Cairns, 2007). Map was provided by the project Hotspot Ecosystem Research and Man’s Impact on European Seas (HERMIONE).
The collection of new samples during the deep-sea campaign BobECo (Autumn 2011, R/V Pourquoi Pas?, IFREMER, French-Irish margins) provided additional specimens for the deep-sea species complex H. dedritifera and permitted the analysis of distantly related sponge species (that is, Mycale sp.) and ambient water samples from the same locations. Samples were collected by the ROV (remotely operated vehicle) Victor in sterile and isolated compartments filled with 0.22 μm filtered seawater before sampling. After collection, individuals were transferred to −80 °C. In some cases, sponge specimens collected in non-sterile boxes were also analyzed after flushing on deck with 0.22 μm filtered seawater to eliminate loosely attached microorganisms. Two liter seawater samples were collected in close proximity to the sampled sponges in sterile water bags and filtered through individual 0.22 μm sterivex filters (Durapore, Millipore, MA, USA). Two replicate filters were processed per sterile water bag; one was stored directly at −80 °C and the other in 96% ethanol. Some locations were explored during independent dives. Only one ethanol-fixed filter was collected from a second dive on ‘Petite Sole’. These different storage conditions provided an opportunity to assess how sample storage impacts microbial communities composition and structure. Total genomic DNA was extracted using a MoBio PowerPlant DNA Isolation Kit following the manufacturer’s instructions (MO BIO Laboratories, Carlsbad, CA, USA).
Amplification of the 5′ partition (that is, the standard Folmer barcoding fragment) of the cytochrome c oxidase subunit I, sequences alignment and phylogenetic analyses were done as in the study by Reveillaud et al. (2010). Hexadella sequences were checked for similarities with the haplotypes from the above study. Mycale sp. sequences were verified for their poriferan origin by BLAST searches against the GenBank database (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Bacterial community characterization using Illumina paired-end tag sequencing
The V6 region of the 16s rRNA gene (ca. 60 nucleotides) was PCR amplified in triplicate for each sample using a combination of four forward primers 967F (5′-CTAACCGANGAACCTYACC-3′; 5′-ATACGCGARGAACCTTACC-3′; 5′-CNACGCGAAGAACCTTANC-3′; 5′-CAACGCGMARAACCTTACC-3′) and one degenerate reverse primer 1046R 5′-CGACRRCCATGCANCACCT-3′, fused to barcodes compatible with the Illumina HiSeq1000 sequencing platform bridge adapters as described in the study by Eren et al., (2013b). The amplicon libraries in this study were sequenced in a single run on two different Illumina HiSeq lanes for technological sequencing replicates. Although the two lanes were merged together for the data analysis, the data sets were also analyzed separately for each lane in order to assess technical variation among technical replicates (data not shown).
Quality filtering and sequence analysis
Sequence quality trimming and filtering relied upon perfect identity of paired-end read overlaps (Eren et al., 2013b). This approach generates very high-quality reads by eliminating the majority of sequencing errors. The VAMPS database (https://vamps.mbl.edu) provides public access to quality-filtered V6 reads under the project name JCR_SPO_Bv6 (Hexadella Microbiome). This study only included 58 samples from seawater and specimens of verified poriferan origin (see Table 1). The QIIME 1.5 pipeline (Caporaso et al., 2010) was used for rarefaction and clustering analyses. Reads were clustered into operational taxonomic units (OTUs) at 97% sequence identity level (which is equivalent to one nt difference between ∼62 nt long V6 reads) using USEARCH (Edgar, 2010), with the minimum word length parameter set to 30. The Global Assignment of Sequence Taxonomy (GAST, Huseet al., 2008) assigned taxonomy to reads using the SILVA 111 database (Quast et al., 2013). The sequencing depth for each sample relied upon a resampling of the OTU table to the smallest sequencing depth, with a total of 100 000 bacterial reads per sample for the beta-diversity analyses. An additional rarefaction at 400 000 reads per sample (excluding samples H20D, 10ION, H18, 44ION and D14ROS) was tested to confirm the patterns (data not shown). Two distinct indices (binary Jaccard: presence–absence and Morisita-Horn: relative abundance) estimated dissimilarity in pairwise comparisons of bacterial communities. A data set filtered for OTUs with fewer than 1000 occurrences across all samples (92 277 OTUs represented by 6 252 406 reads) was analyzed separately in order to explore host-specificity pattern among the rare members of the sponge microbiome. Normalization of sequencing depth per sample employed a resampling of the OTU table to a total of 6000 bacterial reads per sample (that is, the smallest sequencing depth per sample) for the beta-diversity analyses. An additional rarefaction at 40 000 reads per sample (excluding 12 samples, see Supplementary Material) was tested to confirm the patterns.
Oligotyping
The oligotyping computational method (Eren et al., 2011, 2013a) further explored potential host-specificity patterns using subtle nucleotide variation in closely related Hexadella microbiota. From the entire data set, we submitted 2 112 927 high-quality Illumina V6 fragments that GAST identified as Nitrospira to the oligotyping pipeline version 0.6 (available from http://oligotyping.org). A total of 22 nucleotide positions (that is, entropy components) identified oligotypes among reads following the initial entropy and oligotyping analysis. To reduce noise, only oligotypes that minimally occurred at least 100 times across all samples were included.
LEfSe
LEfSe analysis (Segata et al., 2011) explored the potential presence of taxonomic groups that can serve as biomarkers for different classes. Statistically significant groups are reported with high LDA (Linear Discriminant Analysis) scores, which characterize the degree of consistency in relative abundance between features (taxonomic groups) together with their effect relevance (that is, effect size) in each class. The Supplementary Material describes other statistical analyses for data presented therein.
Results
Diversity of microbial communities and taxonomic richness
Stringent quality filtering of 129 285 979 Illumina reads retained 82 560 660 high-quality sequences from sponge and seawater samples for downstream analyses. The number of reads per sample ranged from 101 754 to 6 345 182 (mean 1 423 459+/−935 186). Cluster analysis at 97% identified 96 279 OTUs across the entire data set including 29 457 different 97% OTUs for a single seawater sample (W16; see Supplementary Table 1) and between 9667 and 48 851 for different sponge species. Rarefaction curves for individual samples (Supplementary Figure 1) revealed a long tail distribution where low-abundance OTUs, that is, rare taxa (for example, below 0.01%) represented most of the diversity in the sample, whereas fewer than 100 OTUs accounted for highly abundant OTUs (for example, above 1%). Supplementary Figure 2 and Supplementary Tables 2A and B, 3 and 4 report data trends not specific to the tested hypothesis including estimates of taxonomic richness.
Bacterial community composition and structure
The sponge-associated bacterial communities showed a distinct membership from those in the water column and members of each sponge species shared a microbiome composition that differed from the microbiomes of other sponge species in this study (Figure 2, Supplementary Figure 3A). The Mycale samples displayed a distinct bacterial community from the Hexadella hosts with no overlap between the overall community compositions of the different Hexadella sponge species. Despite some inter-individual variation, the data indicate that geographically distant deep-sea populations of the same sponge species hold similar bacterial community composition. For example, the bacterial community composition of the deep-sea species H. dedritifera from the Bay of Biscay to the Greenland Sea had similar microbial community compositions, and H. cf. dedritifera from the Mediterranean Sea to Ireland harbored similar bacterial communities (Supplementary Figure 3A). Further, sponge species (that is, H. dedritifera versus H. cf. dedritifera) had a significant influence (PERMANOVA (permutational multivariate analysis of variance), P=0.018) on bacterial community composition from the same location (that is, Rockall, see Supplementary Table 5). These results further show strong sponge-specific and species-specific community membership for sponge microbiomes. A filtered data set, discarding OTUs with more than 1000 reads, revealed similar patterns (See Supplementary Figures 4A and B), where host-specific bacterial community relationships extend to rare members of the sponge microbiome. The structure of the sponge-associated bacterial communities, examined using the Morisita-Horn abundance-weighted diversity index revealed similar patterns, with a number of outliers (See Supplementary Figure 3B).

Dendogram of microbial communities (UPGMA:Unweighted Pair Group Method with Arithmetic Mean) using Jaccard. The scale bar represents binary Jaccard distances.
Bacterial community in relation to Hexadella phylogeny and the environment
When analyzed in the context of the Hexadella phylogeny, the relationships between bacterial communities lacked congruency with the known evolutionary relationships of the hosts (see Supplementary Figure 5 and the Mantel test, P=0.004). For instance, the H. pruvoti sponges (in bright green) do not contain more similar microbial communities (in both incidence and abundance, data not shown) to the closely related H. cf. dedritifera (in red) than to species from the phylogenetically more distant H. dedritifera (in orange) species. This indicates that the Hexadella microbiota did not solely diversify through parental inheritance.
To examine the extent of the sponge-specific bacterial community, we examined the shared and the species-specific OTUs (defined at the 97% clustering level across all samples) for each biome (that is, Hexadella, Mycale and seawater). Among 96 279 OTUs, sponges and seawater shared 37 323 OTUs (38%), 49 186 OTUs (51%) were specific for sponges (found exclusively in sponges), whereas 9770 (10%) occurred only in seawater (seawater specific; see Figure 3a). We also assessed the shared and specific OTUs for each biome from 27 sponge and seawater samples collected from the same geographic locations (that is, French and Irish margins), including Hexadella cf. dedritifera (11 samples), Mycale species (5 samples) and ambient seawater samples (11 samples). This data set (hereafter referred to as the ‘restricted data set’, highlighted in bold in Table 1) contained 69 627 OTUs that we further investigated in order to determine the fraction of OTUs that is shared between sympatric sponge species and surrounding seawater. In this more conservative approach, the sponge-specific fraction was much lower (32%), whereas the shared fraction and the seawater specific fractions were higher (42% and 25%, respectively; Figure 3b).

Venn diagram of the shared and specific bacterial OTUs across the Hexadella, Mycale and seawater biomes. (a) Analysis of 58 sponge and seawater samples. (b) Analysis of 27 sponge and seawater samples from the same French and Irish locations.
Potential biomarker taxa were examined using LEfSe, which identified 33 taxonomic groups distinguishing sponge and seawater communities. These groups included 5 Nitrospirae biomarkers for the sponge class, and 26 distinct Proteobacteria that were divided into 24 groups for the seawater class and 2 for the sponge class (Supplementary Figure 6), all with a LDA larger than 4.
Oligotyping of the genus Nitrospira
The genus Nitrospira, generally comprised of nitrite-oxidizing bacteria, represented 0.14% and 12.91% of the 16S rRNA genes in most Hexadella microbiomes, but fewer than 0.058% of the sequences in the Mycale sp. and seawater specimens (Figure 4). Oligotyping analysis of sequences belonging to this group identified 261 oligotypes, represented by 2 070 103 of 2 112 927 (97.97%) Nitrospira reads. Oligotype distribution clearly differed in Hexadella spp versus Mycale and the seawater (Figure 4) communities. The analysis also shows that some conspecifics Hexadella retain very similar oligotypes (for example, in H. dedritifera or in Hexadella sp). Multidimensional scaling analysis using both Binary Jaccard (Supplementary Figure 7) and Morisita–Horn (data not shown) confirmed that oligotypes for members of a particular species were more similar to one another than to those of other species. These results also show that individuals of deep-sea species, such as H. dedritifera, grouped together at the genus level despite spanning a wide geographical distribution from the Bay of Biscay to the Greenland Sea.

Top: Stack-bar figure for Nitrospira oligotypes among the seven Hexadella species, Mycale and seawater samples. Bottom: corresponding percent abundance of Nitrospira per sample (Square root transformed).
Nitrospira oligotypes within groups containing at least five samples (that is, H. dedritifera, H. cf. dedritifera, Mycale sp. and seawater background) were analyzed using LEfSe for biomarker identification. Two Nitrospira oligotypes were found as biomarkers for the class of Hexadella cf. dedritifera and for the classes of Mycale sp. and seawater, respectively, with a LDA larger than 5. The most dominant Nitrospira oligotype GTCCCAATGG_TAA_TCCGCAG (oligotype A) represented by 852 546 reads was highly abundant in H. cf. dedritifera, but relatively rare in H. dedritifera, Mycale sp. and seawater. Oligotype B (ACCTTGACGG_TGC_GTCTTCT, represented by 5764 reads) was more abundant in Mycale sp. and seawater than in Hexadella spp (Figure 5). These results suggest that some Nitrospira sequences are enriched in certain Hexadella species, whereas others are not enriched in the Hexadella sponge niche.

Biomarker detection analysis (LEfSe) on Nitrospira oligotypes of H. dedritifera, H. cf. dedritifera, Mycale sp. and seawater communities. LEfSe identified two biomarkers (A and B) oligotypes that are differentially abundant and with a LDA larger than 5 in the classes of H. cf. dedritifera and of Mycale sp. and seawater, respectively. The mean and median relative abundance of the biomarker oligotypes are indicated with solid and dashed lines, respectively.
Discussion
This study describes the first in-depth investigation of bacterial communities in closely related and cryptic sponges species across a wide array of geographic and bathymetric localities using high-throughput sequencing. Samples were collected from shallow to deep-sea sites (15–960 m) along the Mediterranean Sea up to the Greenland Sea. Together with a recently resolved sponge phylogeny generated from nuclear and mitochondrial markers (Reveillaud et al., 2010), this sample collection provides the most extensive framework for thoroughly characterizing host-bacterial specificity.
Sponge-specific and species-specific symbiont associations
Our analyses confirmed a clear sponge-specific and species-specific signature of bacterial community composition, in agreement with previous studies employing less sequencing coverage (Webster et al., 2010; Erwin et al., 2011, 2012; Lee et al., 2011; Webster and Taylor, 2012). Sponge samples stored in a variety of ways also clustered by species, suggesting the signal override the preservatives, as previously shown (Simister et al., 2011). However, this study provides the first evidence for host-specific patterns that extends to rare members of the sponge microbiome. Rare organisms (those that were never found above 0.01% relative abundance in any sample) accounted for more than 90% of the distinct OTUs. Despite this high diversity of rare bacterial members, beta-diversity analysis using indices taking into account solely the presence and absence of taxa (that is, binary Jaccard) demonstrated strong species-specific community membership. Further, similar patterns of host species-specificity were detected among members of the sponge microbiome represented by less than 0.001% of the reads only (restricted to OTUs with less than 1000 reads across all samples). Sequencing and PCR errors can potentially contribute to OTU inflation, notably in the rare biosphere, and evaluating the relevance of singletons and doubletons remains an open research challenge in V6 amplicon sequencing. The strongest evidence that a unique sequence in a sample is of biological origin occurs when this sequence is observed across multiple samples. Here, only 33 OTUs were observed fewer than two times (that is, singletons) and 18 OTUs were found as doubletons. Therefore, most unique sequences in any given sample occurred in at least another sample generated by independent PCR reactions. These results provide evidence that the observed rare taxa in this data set are mostly real. Our data suggest that specific host–bacteria associations extend beyond the dominant taxa into the rare biosphere.
This study provides the first evidence that deep-sea sponge species separated by great geographical distances along the European continental margins (for example, Hexadella cf. dedritifera, occurring from the Mediterranean Sea to the Irish Margins, separated by ca. 5000 km) share similar bacterial community composition. This could reflect post-glacial currents occurring along the European margins (that is, the Mediterranean Outflow Water). Connectivity patterns among cold-water coral faunal assemblages were observed using paleoecological U/Th datings of corals (Schröder-Ritzrau et al., 2005; Roberts et al., 2006) and phylogenetic analysis of sponge species (see Reveillaud et al., 2010). Interestingly, the species of the recently highlighted complex of cryptic deep-sea species (that is, H. dedritifera, H. cf. dedritifera and Hexadella sp) harbored a discrete bacterial community. Similarly, the recently identified shallow-water species H. pruvoti, H. crypta, H. topsenti and H. racovitzai showed divergent associated bacterial communities. Differential metabolic patterns occur within each of these four shallow-water sponge groups (Reveillaud et al., 2012), and although no direct inference can be made concerning the actual producers of secondary metabolic compounds in these sponge-microbe associations, divergent bacterial communities could explain different secondary metabolites profiles for the different Hexadella species.
Specific host–Nitrospira associations
The genus Nitrospira has a key role in nitrogen cycling in sponges (Hentschel et al., 2012; Radax et al., 2012). Our analysis shows that Nitrospira is a dominant taxa as well as a biomarker group for the sponge microbiome. A significantly higher abundance of Nitrospira reads were observed in Hexadella spp versus in Mycale sp. and in the seawater samples, suggesting that these nitrite-oxidizing bacteria may serve a key role in removing ammonia that accumulates in Hexadella sp. In contrast, the restricted amount of Nitrospira in Mycale suggests that another member of the community may perform nitrification in this species or that other specific pathways (for example, sulfate reduction, denitrification or anammox, described in species experiencing anaerobiosis, see Hoffmann et al., 2009) might provide greater fitness to Mycale sp. The relative abundance of Nitrospira in Hexadella spp compared with other sponge species (that is, Mycale) suggests that this particular genera specifically occurs in certain sponge species. Future analyses including metagenomic and metatranscriptomic work will allow for investigation of whether or not metabolic interdependencies determine the stability of host–Nitrospira associations.
Oligotype analysis of sequences belonging to this group indicated that very closely related members of Nitrospira present distinct patterns of diversity and relative abundance among the different Hexadella spp, Mycale and seawater biomes (Figure 4 and Supplementary Figure 7). Although previous work reported host–bacteria specificity at the genus level in cyanobacteria (Thacker and Starnes, 2003), our study revealed that some Nitrospira variants are differentially enriched in closely related sponge species, whereas others are present in Mycale sp. or seawater but not enriched in the Hexadella sponge niches. The LEfSe analysis on Nitrospira oligotypes from H. dedritifera, H. cf. dedritifera, Mycale and seawater samples revealed two Nitrospira oligotypes that were differentially abundant in H. cf. dedritifera and in the Mycale and seawater classes, respectively (Figure 5). These results suggest the selective retention and/or enrichment of strains within the genus Nitrospira in sponges. Our data are consistent with the results from a recent sponge bacteria comparative metagenomic study by Thomas et al. (2010), which showed an abundance of functional genomic signatures, such as symbiotic repeat proteins for the bacterial community. This observation suggests that the sponge can discriminate between food and resident bacteria and that metabolic interdependencies may exist between the host and its associated taxa.
The oligotyping data demonstrate the power of ultra-deep sequencing technologies to both enable identification of unknown reservoirs of bacterial diversity at very fine phylogenetic levels (that is, with the genus Nitrospira) as well as to improve our understanding of the host-specific associations. The results reveal under-estimated diversity within an important member of the sponge microbiome and that host species-specific symbionts exist at the genus level within dominant and rare members of the sponge microbiome. There is a high degree of host-taxa specificity at fine phylogenetic scales, which cannot be detected with coarser 16S rRNA gene clustering or taxonomic assignments.
Vertical and horizontal mechanisms for transmission of sponge–bacteria
On one hand, our results suggest that a high proportion (32%) of symbionts pass from the parents to the offspring and never appear in the seawater, consistent with previous studies (see Schmitt et al., 2012; Webster et al., 2010). On the other hand, our data show a non-negligible proportion (38%) of shared OTUs between sponge and seawater. This frequency was as high as 42% when analyzing only sympatric samples from the same French and Irish locations. These data support the recent study of Taylor et al. (2012), which showed that many microbial taxa found in sponges also occur at very low abundance in the seawater and might serve as seed bank for sponges. Although the Illumina sequencing allowed recovery of a high number of OTUs, deeper sequencing of rRNA amplicons from the seawater (or from sediments, see Schöttner et al., 2013) rare biosphere might reveal additional shared community membership. Overall, ultra-deep sequencing technology demonstrated that both shallow and deep-sea sponge-associated bacterial communities are highly diverse and controlled by both host and environmental-related factors.
Despite the interplay of both the host and the environment, our results indicate the existence of strong ecological and/or evolutionary factors driving these diverse but stable sponge-associated bacteria, including both dominant and rare bacterial members. The exact evolutionary mechanisms and timescales used by sponges and symbionts to preserve these remarkably stable associations remain unknown, but the close associations within the rare biosphere are a particularly exciting finding. In agreement with previous studies (Schmitt et al., 2012), the strict core community (defined as presence in all sponges) was extremely small and consisted of only ca. 1% of the total bacterial community. In contrast, the sponge species-specific community (defined as presence in only a single sponge species) consisted of ca. 30% of the total community. Different sponge species likely represent different ecological niches for bacteria, each acquiring and selectively maintaining a specific microbial community that is acquired both from the environment and from the parents (Webster et al., 2010). Sponge bacterial consortia could date back to the beginning of the animal evolution (Taylor et al., 2007b), and therefore some taxa may be associated with their host possibly over evolutionarily extended periods of time. However, such specialization might not be just ancient. For instance, the large diversity of Nitrospira variants, such as within H. dedritifera, might provide to the host a large and diverse repertoire of genes, which could rapidly expand its metabolic potential, and thus increasing both its ecological plasticity and adaptation to environmental change. Patterns of host-specificity at very fine molecular resolution, such as within the dominant genus Nitrospira, suggest a phylogenetic specificity that warrants further investigation at the functional level. Metagenomic library construction will allow the recovery of functional genes and operons and improve our understanding of the physiological roles of these diverse but highly specialized sponge-associated microbiota.
In conclusion, we exploited the power of ultra-deep sequencing and a well-resolved host phylogeny to characterize host-specificity patterns in sponge microbiota. The results shed light on an exceptional diversity of dominant to low-abundance taxa, including within lineages with known functional importance (that is, Nitrospira). This study demonstrates the stability and specificity of bacterial communities along unprecedentedly wide bathymetric gradients and large geographic areas within both the dominant and rare bacterial biosphere. Oligotyping analysis demonstrates for the first time that closely related sponge species have differential enrichment preferences for closely related Nitropsira variants. It is clear that sponges must use specific mechanisms to selectively maintain bacteria seeded from the seawater and/or transmitted from the parents to offspring. Whether ‘everything is everywhere’ or not, the sponge selects.
References
Cairns SD . (2007). Deep-water corals: an overview with special reference to diversity and distribution of deep-water scleractinian corals. Bull Mar Sci 81: 311–322.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Cebrian E, Uriz MJ, Garrabou J, Ballesteros E . (2011). Sponge mass mortalities in a warming Mediterranean Sea: are cyanobacteria-harboring species worse off? PLoS One 6: e20211.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG et al. (2013a). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol 4: 1111–1119.
Eren AM, Vineis JH, Morrison HG, Sogin ML . (2013b). A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One 8: e66643.
Eren AM, Zozaya M, Taylor CM, Dowd SE, Martin DH, Ferris MJ . (2011). Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PLoS One 6: e26732.
Erwin PM, Olson JB, Thacker RW . (2011). Phylogenetic diversity, host-specificity and community profiling of sponge-associated bacteria in the northern Gulf of Mexico. PLoS One 6: e26806.
Erwin PM, Pita L, López-Legentil S, Turon X . (2012). Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl Environ Microbiol 78: 7358–7368.
Gootenberg DB, Turnbaugh PJ . (2011). Companion animals symposium: humanized animal models of the microbiome. J Anim Sci 89: 1531–1537.
Hentschel U, Horn M, Friedrich AB, Wagner M, Moore BS . (2002). Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microbiol 68: 4431–4440.
Hentschel U, Piel J, Degnan SM, Taylor MW . (2012). Genomic insights into the marine sponge microbiome. Nat Rev Microbiol 10: 641–654.
Hentschel U, Usher KM, Taylor MW . (2006). Marine sponges as microbial fermenters. FEMS Microbiol Ecol 55: 167–177.
Hoffmann F, Radax R, Woebken D, Holtappels M, Lavik G, Rapp HT et al. (2009). Complex nitrogen cycling in the sponge Geodia barretti. Environ Microbiol 11: 2228–2243.
Huse SM, Dethlefsen L, Ja Huber, Mark Welch D, Welch DM, Relman Da et al. (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genetics 4: e1000255.
Lee OO, Wang Y, Yang J, Lafi FF, Al-Suwailem A, Qian PY . (2011). Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J 5: 650–664.
McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Loso T, Douglas AE et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci USA 110: 3229–3236.
Proctor LM . (2011). The Human Microbiome Project in 2011 and beyond. Cell Host Microbe 10: 287–291.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res 41: D590–D596.
Radax R, Hoffmann F, Rapp HT, Leininger S, Schleper C . (2012). Ammonia-oxidizing archaea as main drivers of nitrification in cold-water sponges. Environ Microbiol 14: 909–923.
Reveillaud J, Allewaert C, Pérez T, Vacelet J, Banaigs B, Vanreusel A . (2012). Relevance of an integrative approach for taxonomic revision in sponge taxa: case study of the shallow-water Atlanto-Mediterranean Hexadella species (Porifera:Ianthellidae:Verongida). Invertebr Syst 26: 230–248.
Reveillaud J, Remerie T, van Soest R, Erpenbeck D, Cárdenas P, Derycke S et al. (2010). Species boundaries and phylogenetic relationships between Atlanto-Mediterranean shallow-water and deep-sea coral associated Hexadella species (Porifera, Ianthellidae). Mol Phylogenet Evol 56: 104–114.
Roberts JM, Wheeler AJ, Freiwald A . (2006). Reefs of the deep: the biology and geology of cold-water coral ecosystems. Science 312: 543–547.
Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 6: 564–576.
Schröder-Ritzrau A, Freiwald A, Mangini A . (2005). U/Th dating of deep-water corals from the eastern North Atlantic and the western Mediterranean Sea. In: Freiwald A, Roberts JM (eds) Cold-Water Corals and Ecosystems. Springer: Heidelberg, Germany, pp 157–172.
Schöttner S, Hoffmann F, Cárdenas P, Rapp HT, Boetius A, Ramette A . (2013). Relationships between host phylogeny, host type and bacterial community diversity in cold-water coral reef sponges. PLoS One 8: e55505.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12: R60.
Selvin J, Shanmugha PS, Seghal KG, Thangavelu T, Sapna BN . (2009). Sponge-associated marine bacteria as indicators of heavy metal pollution. Microbiol Res 164: 352–363.
Simister RL, Schmitt S, Taylor MW . (2011). Evaluating methods for the preservation and extraction of DNA and RNA for analysis of microbial communities in marine sponges. J Exp Mar Biol Ecol 397: 38–43.
Taylor MW, Radax R, Steger D, Wagner M . (2007a). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microb Mol Biol Rev 71: 295–347.
Taylor MW, Thacker RW, Hentschel U . (2007b). Evolutionary insights from sponges. Science 316: 1854–1855.
Taylor MW, Tsai P, Simister RL, Deines P, Botte E, Ericson G et al. (2012). ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J 7: 438–443.
Thacker W, Starnes S . (2003). Host specificity of the symbiotic cyanobacterium Oscillatoria spongeliae in marine sponges, Dysidea spp. Mar Biol 142: 643–648.
Thomas T, Rusch D, DeMaere MZ, Yung PY, Lewis M, Halpern A et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J 4: 1557–1567.
Webster NS, Cobb RE, Negri AP . (2008). Temperature thresholds for bacterial symbiosis with a sponge. ISME J 2: 830–842.
Webster NS, Taylor MW, Behnam F, Lücker S, Rattei T, Whalan S et al. (2010). Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol 12: 2070–2082.
Webster NS, Taylor MW . (2012). Marine sponges and their microbial symbionts: love and other relationships. Environ Microbiol 14: 335–346.
Webster NS, Webb RI, Ridd MJ, Hill RT, Negri AP . (2001). The effects of copper on the microbial community of a coral reef sponge. Environ Microbiol 3: 19–31.
Acknowledgements
We thank the captains and crew of R/Vs Belgica, Pelagia, Pourquoi Pas?, G.O. Sars, Polarstern and Minibex. We thank Rob van Soest, Thierry Perez, Jean Vacelet and Joana Xavier for providing samples; Sophie Arnaud-Haonlt, Olivier Mouchel, Valérie Cueff-Gauchard and Inge van den Beld for logistic assistance in collecting and shipping samples; Nicola Segata and Sheri Simmons for helpful discussions on LEfSe analysis; Hilary Morrison, Joseph Vineis, and Sharon Grim for assistance with sequencing; Richard Fox for support with the use of MBL super-computers; Nuria Fernandez and Christopher Algar for guidance with R scripts. Two anonymous reviewers are acknowledged for constructive comments, which significantly improved the earlier drafts of the manuscript. Funding was provided by the European Community’s Seventh Framework Programme (FP7/2007–2013) under the HERMIONE project, grant agreement No. 226354.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Reveillaud, J., Maignien, L., Eren, A. et al. Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J 8, 1198–1209 (2014). https://doi.org/10.1038/ismej.2013.227
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.227
Keywords
This article is cited by
-
Sponges and their prokaryotic communities sampled from a remote karst ecosystem
Marine Biodiversity (2024)
-
Future ocean conditions induce necrosis, microbial dysbiosis and nutrient cycling imbalance in the reef sponge Stylissa flabelliformis
ISME Communications (2023)
-
New Negombata species discovered: latrunculin mystery solved
Coral Reefs (2023)
-
Oceanographic setting influences the prokaryotic community and metabolome in deep-sea sponges
Scientific Reports (2022)
-
Unravelling the interplay of ecological processes structuring the bacterial rare biosphere
ISME Communications (2022)
