
Abstract
Cellulose is the most abundant biopolymer on Earth. Optimising energy recovery from this renewable but recalcitrant material is a key issue. The metaproteome expressed by thermophilic communities during cellulose anaerobic digestion was investigated in microcosms. By multiplying the analytical replicates (65 protein fractions analysed by MS/MS) and relying solely on public protein databases, more than 500 non-redundant protein functions were identified. The taxonomic community structure as inferred from the metaproteomic data set was in good overall agreement with 16S rRNA gene tag pyrosequencing and fluorescent in situ hybridisation analyses. Numerous functions related to cellulose and hemicellulose hydrolysis and fermentation catalysed by bacteria related to Caldicellulosiruptor spp. and Clostridium thermocellum were retrieved, indicating their key role in the cellulose-degradation process and also suggesting their complementary action. Despite the abundance of acetate as a major fermentation product, key methanogenesis enzymes from the acetoclastic pathway were not detected. In contrast, enzymes from the hydrogenotrophic pathway affiliated to Methanothermobacter were almost exclusively identified for methanogenesis, suggesting a syntrophic acetate oxidation process coupled to hydrogenotrophic methanogenesis. Isotopic analyses confirmed the high dominance of the hydrogenotrophic methanogenesis. Very surprising was the identification of an abundant proteolytic activity from Coprothermobacter proteolyticus strains, probably acting as scavenger and/or predator performing proteolysis and fermentation. Metaproteomics thus appeared as an efficient tool to unravel and characterise metabolic networks as well as ecological interactions during methanisation bioprocesses. More generally, metaproteomics provides direct functional insights at a limited cost, and its attractiveness should increase in the future as sequence databases are growing exponentially.
Similar content being viewed by others


Introduction
Lignocellulosic materials including paper, textile, wood, yard trimmings and crop straws are dominant in municipal solid waste (MSW), agricultural waste and energy crops. Lignocellulose is the most abundant biochemical renewable energy source on Earth, because the bioenergy it contains can be recovered by cost-effective and robust anaerobic digestion technologies that are now widely applied. These processes involve sophisticated self-assembled and largely uncultured microbial communities comprising tens to hundreds of operational taxonomy units (Chouari et al., 2005; Krause et al., 2008; Kröber et al., 2009; Riviere et al., 2009). However, various biotechnological barriers still hinder a fully optimised process operation. Major issues include the polysaccharide hydrolysis efficiency, the methanogenesis stability and the sensitivity to inhibitions and disturbances (Chen et al., 2008; Ward et al., 2008; Ganidi et al., 2009; Holm-Nielsen et al., 2009). To better pilot anaerobic bioprocesses, additional functional insight into the catalysis of complex organic substrate digestion by the anaerobic microbial communities is required.
Significant progress has been achieved over the last few years. Fluorescent in situ hybridisation (FISH) approaches contributed to the identification of cellulolytic bacteria in various anaerobic environments (O’Sullivan et al., 2007). Isotopic labelled substrates were exploited to identify the functional microbial groups catalysing the methanisation of cellulose using stable isotope probing (Li et al., 2009). Several metagenomic studies shed light on the identity of the functional groups and provided an extended catalogue of the catalytic potential (Krause et al., 2008; Schlüter et al., 2008; Jaenicke et al., 2011; Rademacher et al., 2012). However, these approaches do not generate direct information on the expressed genes and associated metabolic processes. Recently, metatranscriptome sequencing provided insight into the metabolically active communities of a mesophilic biogas plant (Zakrzewski et al., 2012). Two metaproteomic approaches were implemented on similar anaerobic systems, and the feasibility of such approaches was proved (Abram et al., 2011; Hanreich et al., 2012), and more specifically on the complex matrix of anaerobic lignocellulose-degrading communities (Hanreich et al., 2012). Both based on a two-dimensional gel separation, they enabled the identification of a few dozen functions in the case of psychrophilic anaerobic digestion of glucose-fed wastewater (Abram et al., 2011) and of a dozen functions for methanisation of agricultural biomass in thermophilic conditions (Hanreich et al., 2012).
As thermophilic methanisation is currently emerging as a promising process (van Lier et al., 2001), we investigated the metaproteome obtained from a microcosm containing office paper and inoculated with stabilised digestate from a thermophilic MSW anaerobic digester, with the objective of achieving a comprehensive view. To increase the identification depth, a combination of various separation techniques was employed, and a high number of protein fractions were analysed. The possibility of obtaining valuable information based solely on public protein databases was questioned. For this, identification results using the UniprotKB database were compared with analyses of the microcosm generated independently by a polyphasic approach (16S rRNA gene pyrosequencing, FISH, stable isotopic fractioning signatures of methanogenesis processes). Encouragingly, more than 500 non-redundant protein functions were identified, and the complementary approaches were in overall good agreement.
After presenting an overview of the metaproteomic data set and assessing its validity, functional insights are presented with a focus on each methanisation step (hydrolysis, fermentation, acetogenesis and methanogenesis) and on unexpectedly high proteolytic activities.
Materials and methods
Anaerobic incubations
Sludge was sampled from a 21-m3 thermophilic anaerobic industrial pilot digester located in France, fed with the organic fraction of MSW. The sample was sieved, stabilised at 55 °C, centrifuged at 13 100 g at 4 °C, aliquoted and stored at −80 °C to serve as inoculum. In each of five 1-l bottle (replicates A–E), 5 g unprinted office paper, 10 g inoculum (wet mass) and 500 g Biochemical Methane Potential buffer (EN-ISO-11734, 1998) were added (see also Supplementary Materials Section S0). Three similar microcosms without paper were set up as control. The bottles were rubber-sealed, the headspace was purged with N2 and all microcosms were incubated in anaerobic and thermophilic conditions (55±0.5 °C).
Chemical analyses
The degradation dynamics was assessed by measuring the biogas production and composition, pH, concentrations of total organic carbon, total inorganic carbon and volatile fatty acids in the liquid phase. The biogas isotopic composition was analysed by determining δ13CH4 and δ13CO2 values and calculating the apparent fractionation factor αC (Qu et al., 2009). Biogas production and composition were assessed as described in Qu et al. (2009). The detected gas included CO2, CH4, H2S, N2, H2 and O2. The liquid samples were recovered with a syringe and needle through the rubber septum. They were centrifuged briefly at maximum speed in a bench centrifuge to separate the liquid phase from the cell-containing pellet. The pH was measured on the liquid phase just after sampling. The supernatants and pellets were stored separately at −80 °C for further chemical and biological analyses.
Protein preparation and mass spectrometry analyses
Proteins were extracted and purified from 50 ml samples of the paper anaerobic incubation (replicate A, day 60) using a protocol from Wilmes and Bond (2004), modified to handle samples from a MSW digester containing much debris. Briefly, cells were disrupted by bead-beating in the lysis buffer, and the proteins were purified from the obtained supernatant by trichloroacetic acid–acetone precipitation. Protein concentrations were assessed using the 2-D Quant Kit (GE Healthcare, Aulnay sous Bois, France) and the High Sensitivity Protein 250 Kit (Agilent, Les Ulis, France). The purified proteins were further processed for subsequent MS/MS analyses according to three different strategies detailed below. For every strategy, fixation of each fraction in SDS-PAGE gel (NuPAGE Novex 4–12% Bis-Tris Gel 1.0 mm; Invitrogen, Saint Aubin, France) was the last step, with or without prior separation. Strategy 1 was the separation according to the molecular weight, resulting in 26 fractions: a protein aliquot was migrated by SDS-PAGE and the gel lane was cut into 26 sections. Strategy 2, performed in three technical replicates, was the separation into 12 fractions according to the pI, resulting in a total of 36 fractions: for each protein aliquot, 12 liquid fractions were generated by off-gel isoelectric focusing (OFFGEL-IEF, Agilent 3100 OFFGEL Fractionator, low-resolution kit, pH 3–10, 12 cm Immobilised pH Gradient strip), and each obtained fraction was fixed into SDS-PAGE gel by a very short-duration migration and gel excision. Strategy 3, performed in three technical replicates, was the absence of separation and generated 3 fractions: the protein aliquot was simply fixed in an SDS-PAGE gel fragment by a very short-duration migration followed by gel excision. As a result, a total of 65 fractions (26+36+3) fixed in SDS-PAGE gel fragments were separately submitted to in-gel tryptic digestion followed by shotgun analyses by nanoLC-MS/MS (LTQ-Orbitrap, Thermo Fisher, Waltham, MA, USA; PAPPSO proteomic platform, INRA, Jouy-en-Josas). The detailed procedures are supplied in the Supplementary Section S1.
Peptide identification and data processing
The mass spectrometry data set produced for each fraction was analysed for peptide and protein identification using X!Tandem software (http://www.thegpm.org/tandem/) and the UniProtKB database with nearly 20 million entries (version January 2012, http://uniprot.org). The 65 X!Tandem result sets were imported into the software Scaffold 2.0 (Proteome Software, Inc., Portland, OR, USA) to combine, compare and validate identified proteins based on peptide and protein probability. The filtering thresholds were the protein probability >95%, at least two unique peptides per protein, and the peptide probability of at least one unique peptide >90%. The potential contaminant proteins, such as keratin and trypsin, were excluded from the analysis.
The obtained identified redundant proteins were grouped according to two different methods: first, non-redundant protein groups were obtained in Scaffold 2.0 based on the presence of shared identified peptides; second, and independently, another grouping was performed based on belonging to shared UniRef clusters with various identity thresholds (for example, 50%, 90% and 100%). The latter methods provided a simple and convenient insight into the taxonomic specificity of the identified redundant proteins. For instance, the case where an identified protein belonged to a UniRef50 cluster containing solely this protein was a strong indication that the taxonomic assignment and the inferred function were very specific because no other closely related protein sequence was present in the database. The possible presence of a signal peptide within protein sequences was analysed with SignalP (Petersen et al., 2011). The detailed procedures are supplied in the Supplementary Materials (Supplementary Section S1).
DNA analyses
DNA was extracted using the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions. A series of 16S rRNA-based techniques were conducted to describe the microbial community, including automated ribosomal intergenic spacer analyses (ARISA, Supplementary Figure S12), 16S rRNA gene pyrosequencing and FISH. Pyrosequences were obtained for the raw digestate directly sampled from the industrial facility, the inoculum (sieved and stabilised digestate, see section Anaerobic incubations), the replicate A (day 0 and 60) and the replicate B (day 60 and 73). They were deposited in the National Center for Biotechnology Information Short Read Archive as BioProject PRJNA182049. More detailed procedures are supplied in the Supplementary Section S2.
Results and Discussion
Office paper degradation dynamics and general overview of the metaproteome data set
Office paper was selected as a model cellulosic substance, because it is a major component in MSW and has a relatively fixed composition of 70% cellulose and 30% hemicellulose. Batch anaerobic incubations of office paper were conducted at 55 °C in five replicated microcosms labelled from A to E. At day 60, the replicate A was used for metaproteomic analyses, whereas replicates B–E were further incubated until day 120 (Supplementary Materials Section S0). The replicates exhibited a good level of reproducibility, and classical degradation trends were observed (Supplementary Figure S2). The rapid onset of a hydrolytic and acidogenic activity led to the accumulation of volatile fatty acid, reaching a concentration of 360 mg carbon per liter at day 18 and mainly corresponding to acetate. This induced a pH decrease from 7.5 to 5.8. After a phase of slow methane production, the methanogenic activity increased gradually from day 20 onwards until it reached a plateau around day 60. At this stage, 62% of the carbon initially introduced as paper had been degraded, and volatile fatty acid concentrations in the liquid phase were low and mainly corresponded to acetate, propionate and lactate.
All metaproteomic analyses were performed on samples from replicate A, day 60: 65 protein sample fractions were prepared by three different separation strategies and analysed by MS/MS (Materials and methods) to favour a sufficient analysis depth (Figure 1a). Strategy 2 based on OFFGEL-IEF generated the highest number of identified non-redundant protein groups (266, 358 and 360 for each technical replicate, respectively), followed by strategy 1 based on SDS-PAGE (212 protein groups) and by strategy 3 (no separation, 54, 90 and 120 protein groups for each technical replicate, respectively) (Supplementary Table S4, Supplementary Figure S5). Both the separation step and the amount of starting protein material appeared as important factors to favour identification (Supplementary Table S3). All the data sets were then combined and analysed together. A significant number of proteins were identified using the UniprotKB database (full content). Among the 7 17 065 spectra obtained, 40 818 (≈6%) were assigned to peptides by X!Tandem search. As a reference, 25% of the spectra are typically assigned when studying 10 μg proteins from a pure culture of Lactococcus lactis (Beganovic et al., 2010). After filtering with Scaffold software, 13 090 peptides corresponding to 2541 potentially redundant proteins were retained. The latter corresponded to 514 non-redundant protein groups and also to 497 distinct UniRef50 clusters (Materials and method). Except for a few protein groups that were highly redundant (for example actin, pyridoxine, RNA polymerase sigma factor and some oxidoreductases), the redundancy was overall limited and distributed throughout the various protein groups.

Overview of the metaproteomics results. (a) Rarefaction curve. Number of identified non-redundant protein groups in function of the number of protein sample fractions included in the analyses. The fractions were generated by several technical replicates and various separation procedures (Materials and methods). A total of 65 protein fractions were included. (b) Distribution of the identified non-redundant protein groups into Clusters of Orthologous Groups. [S] Function unknown, [R] General function prediction only, [Q] Secondary metabolites biosynthesis, transport and catabolism, [P] Inorganic ion transport and metabolism, [I] Lipid transport and metabolism, [H] Coenzyme transport and metabolism, [F] Nucleotide transport and metabolism, [E] Amino acid transport and metabolism, [G] Carbohydrate transport and metabolism, [C] Energy production and conversion, [O] Post-translational modification, protein turnover and chaperon functions, [U] Intracellular trafficking, secretion and vesicular transport, [W] Extracellular structures, [Z] Cytoskeleton, [N] Cell motility, [M] Cell wall/membrane/envelope biogenesis, [T] Signal transduction mechanisms, [V] Defense mechanisms and [Y] Nuclear structure. (c) Taxonomic distribution of the identified non-redundant protein groups. Bold number: number of non-redundant protein groups. Number in parenthesis: number of non-redundant protein groups belonging to UniRef50 clusters specific for the considered taxonomic or functional group (C. proteolyticus species, C. thermocellum species, Caldicellulosiruptor genus, Methanothermobacter genus and other methanogens, respectively) (see Materials and methods for more details about UniRef50). *Putative uncharacterised protein A4XIB5_CALS8 from Caldicellulosiruptor, and/or A7I471_METB6 from Methanogens **RNA polymerase sigma factor A4XHW4_CALS8 from Caldicellulosiruptor and/or A3DDV0_CLOTH from C. thermocellum ***Phenylacetate-CoA ligase A3DD21_CLOTH from C. thermocellum and/or A6VIF0_METM7 from Methanogens.
According to the classification into the 25 Clusters of Orthologous Groups (Figure 1b), the category amino acids [E] seemed unexpectedly abundant, given that the initial substrate was only composed of carbohydrate. Indeed, the dominant categories were related to energy [C], carbohydrate [G] and amino acids [E], together accounting for 46.6% of the 514 non-redundant protein groups. Post-translation [O], translation [J] and coenzyme [H] were the next dominant functional categories, representing 14.6% of the total. Most proteins related to coenzyme [H] were assigned to archaea.
The taxonomic distribution of the identified proteins based on the non-redundant protein groups (Figure 1c, Supplementary Table S17) suggested the presence of a few dominant groups, possibly linked to the thermophilic conditions and the presence of one major substrate for growth (office paper). The retrieved taxa were consistent with the anaerobic thermophilic conditions. Caldicellulosiruptor species (≈41%), Coprothermobacter proteolyticus (≈20%) and Clostridium thermocellum strains (≈16%) were the most represented, suggesting the dominance of members of the order Thermoanaerobacterales followed by Clostridiales. Methanothermobacter thermoautotrophicus was the dominant archaea (≈5%).
Validity and representativeness of the identified proteins
Polyphasic experiments and in silico approaches were implemented to evaluate the validity of the metaproteomic data before developing more detailed biological interpretation. This approach suggested that invaluable functional insight could be gained for most of the dominant microbial groups.
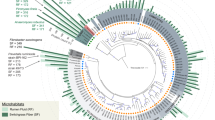
The accuracy of the taxonomic distribution resulting from the metaproteomic data set was evaluated by 16S rRNA gene tag pyrosequencing (Figure 2, Supplementary Figure S13). On the same sample (Figure 2, A60) and on samples from replicate B at similar time points (Figure 2, B60 and B73), the most dominant genera were consistently retrieved (Caldicellulosiruptor, Coprothermobacter and Methanothermobacter), and the dominance of members of the orders Thermoanaerobacterales followed by Clostridiales was confirmed. These dominant strains were not abundant in the inoculum (Figure 2, Inoc, A0; Supplementary Figure S12), and they thus developed in the course of the incubation, which further supports their active role during the office paper digestion. FISH analyses, reflecting the abundance of rRNAs in the specifically targeted cells, were performed for the replicate A at day 60. They provided additional elements of proof for the activity and abundance of the three above-mentioned bacterial genera (Supplementary Figure S11). The genera Gelria (Firmicutes phylum) and Tta-b61 (Firmicutes phylum) were poorly represented in the metaproteomic data set, although they appeared to be of significant importance based on the pyrosequencing results (Figure 2b, operational taxonomic units 489 and 356–383, respectively). These observations were probably mainly linked to the absence of closely related sequenced genomes in the database (Table 1). In addition, in contrast to the metaproteomic analyses, a low proportion of sequences were attributed to C. thermocellum and other Ruminococcaceae cellulolytic strains including Clostridium cellulosi (Figure 2b, A60). One possible reason could be that the classification of Firmicutes is very complex and that the taxonomic assignment results obtained on partial 16S rDNA sequences must be interpreted cautiously (average read lengths ∼300–400 bp). PCR bias on the sample from microcosm A, day 60, could also be an explanation for this discrepancy because abundant related strains were observed in the sample by FISH with probe UCL284 (Supplementary Table S1, Supplementary Figure S11).

Taxonomic distributions obtained by 16S rRNA gene pyrosequencing: (a) with the archaeal primer set and (b) with the bacterial primer set. Dig: collected raw thermophilic digestion sludge; Inoc: thermophilic inoculum (sieved and stabilised digestion sludge); A0, A60, B60 and B73: microcosm and incubation day; * sample also analysed by metaproteomics (A60). For the most abundant groups, the arbitrary operational taxonomic unit (OTU) number is indicated. The cladogram is based on genus frequencies for archaea (a) and on OTU frequencies for bacteria (b). See Supplementary Section S2 and Supplementary Table S1 for more details about the procedures. Rarefaction curves are shown in Supplementary Figure S13.
The identification of peptides and proteins using the full UniprotKB database was highly specific. As pointed out in a study by Denef et al., 2007, similar procedures enable cross-strain identification and avoid most cross-species false-positive hits. Indeed in the present case, the overlap between the dominant microbial groups after protein identification was limited to three non-redundant protein groups (Figure 1c). In addition, 24% of the 427 UniRef50 clusters affiliated to the dominant microbial groups were specific for the concerned group, respectively, Caldicellulosiruptor species, C. proteolyticus, C. thermocellum and methanogenic archaea of various orders (Figure 1c, Supplementary Figure S10). All this confirmed the specificity of the identification procedure, and thus strengthens the validity of the identified functions.
In conclusion, the presence in the database of proteomes from fully sequenced genomes closely related to the sample’s strains together with the specificity of the identification procedure suggested that interesting functional insights could be gained for most abundant groups (C. proteolyticus, C. thermocellum-related strains, Caldicellulosiruptor species, Methanothermobacter species). More limited information was expected for Gelria and Tta-b61 groups, which could have been significantly present in the sample.
Ruminococcaceae strains and Caldicellulosiruptor species are complementary key actors for polysaccharide hydrolysis
Consistent with paper being the main substrate for the incubations, a large number of proteins potentially involved in cellulose and hemicellulose binding and hydrolysis or in oligosaccharide metabolism were identified (Table 2). Ruminococcaceae strains including C. thermocellum, and Caldicellulosiruptor members, appeared as key actors for paper hydrolysis. Moreover, the details of the identified functions suggested a synergetic action of both actors. The former could have been hydrolysing the cellulosic part of the substrate, including the crystalline part, whereas the latter could have been specifically involved in hemicellulose hydrolysis in addition to cellulose hydrolysis.
More specifically, 11 non-redundant protein groups related to polysaccharide hydrolysis were attributed to C. thermocellum. This widely studied thermophilic cellulolytic species is known for efficiently hydrolysing the cellulose, including in its crystalline shape (reviewed in Maki et al., 2009). Its cellulosome has been extensively characterised (reviewed in Bayer et al., 2008), and the presence of dockerin/cohesin domains within a protein is a strong indication that they are cellulosomal subunits. The identified proteins included some major cellulosomal structural and catalytic components. In particular, CelS and CelJ proteins are among the most upregulated enzymes for C. thermocellum Avicel-grown cells compared with cellobiose-grown cells (Gold and Martin, 2007). Hence, even if a limited number of the over 30 cellulosomal proteins from C. thermocellum (Gold and Martin, 2007) were detected, the information was sufficient to confirm the cellulolytic activity of strains closely related to C. thermocellum.
Seven non-redundant protein groups related to polysaccharide hydrolysis were affiliated to the Caldicellulosiruptor genus (Table 2). This taxon contains cellulolytic members, such as C. obsidiansis or C. bescii (formerly Anaerocellum thermophilum). These do not possess cellulosomes, their cellulolytic system being based on secreted multifunctional enzymes; they usually utilise a broad range of plant materials, including crystalline cellulose, cellulose, hemicelllose, starch and pectin, with a very high hydrogen yield (van de Werken et al., 2008; VanFossen et al., 2009; Yang et al., 2009; Hamilton-Brehm et al., 2010; Lochner et al., 2011). Among the non-redundant protein groups identified for Caldicellulosiruptor, four were probably related to hemicellulose degradation (two beta-mannanases UniRef50_Q9KWY5, one acetyl xylan esterase UniRef50_F8F4V8 and one endoxylanase UniRef50_E4Q5G9). This suggested that Caldicellulosiruptor members were also present in the incubations and partly specialised in hemicellulose degradation.
To the best of our knowledge, results on cellulolytic species functions and interactions have not been reported in such detail for cellulose methanisation by complex microbial communities.
Identified proteins related to central carbon metabolism
The identified proteins related to central carbon metabolism (Supplementary Tables S6–S8, Supplementary Figures S7–S9) reinforced the conclusions concerning the major contribution of Caldicellulosiruptor species (see also Supplementary Table S20) and C. thermocellum (see also Supplementary Table S19) to saccharification and fermentation during the incubation, with the former more oriented towards hemicellulolysis compared with the latter, which is more specialised for cellulolysis. In addition, these results highlighted the important contribution of C. proteolyticus as fermenting microorganism (see also Supplementary Table S18) and indicated the possible activity of other non-dominant species. Finally, they suggested that each of these groups produced a distinct combination of metabolic end products and that acetate was produced by most of the identified fermentative bacterial groups.
More specifically, after polysaccharide and oligosaccharide hydrolysis, monosaccharides are channelled to the central catabolic pathways to generate pyruvate. None of the enzymes from the Entner-Doudoroff pathway were detected. In contrast, proteins from the Embden–Meyerhof pathway (glycolysis) corresponding to 24 distinct UniRef50 clusters were identified (Supplementary Table S6, Supplementary Figure S7). Mainly attributed to strains from the C. thermocellum, C. proteolyticus and Caldicellulosiruptor genera, they confirmed the importance of these Gram-positive members of the class Clostridia in the studied community. Remarkably, considering together the identified proteins affiliated to the Caldicellulosiruptor genus, only the phosphofructokinase was not detected; all other nine enzymes from the Embden–Meyerhof pathway were retrieved for this genus. Proteins from the non-oxidative arm of the pentose phosphate pathway were present as well (Supplementary Table S6, Supplementary Figure S7, 10 distinct UniRef50 clusters) and overall attributed to the same three Clostridia taxa. In particular, enzymes processing the xylose—a major building block from hemicellulose—to the pentose phosphate pathway non-oxidative branch were identified and attributed to Caldicellulosiruptor species (Supplementary Table S6, Supplementary Figure S7, for example, xylose isomerase, step 28, and xylulokinase, step 29). This was consistent with the above-mentioned role of Caldicellulosiruptor species in hemicellulolyse.
Proteins associated with pyruvate metabolism and corresponding to 23 distinct UniRef50 clusters were also identified (Supplementary Table S7, Supplementary Figure S8), pyruvate being the glycolytic pathway end product. They were, however, distributed among various taxa, mainly C. thermocellum, C. proteolyticus and Caldicellulosiruptor species (as above), as well as Pelotomaculum thermopropionicum (Clostridia class). The latter is a known anaerobic thermophilic, syntrophic, propionate-oxidising bacterium (Imachi et al., 2002).
Acetyl-CoA and other tricarboxylic acid cycle intermediates are further catalysed into different fermentation products. The identified proteins (Supplementary Table S8, Supplementary Figure S9) corresponded to 21 distinct UniRef50 clusters and were mainly affiliated to Caldicellulosiruptor species and C. proteolyticus, followed by C. thermocellum and Thermosinus carboxydivorans. The latter is an anaerobic thermophilic hydrogen-producing bacterium (Sokolova et al., 2004). According to the identified enzymes, a variety of fermentation products could have been generated. C. thermocellum strains could have been mainly generating lactate, ethanol and acetate as metabolic end products (Supplementary Table S8, (11), (13), (17)). Caldicellulosiruptor species could have been producing mainly lactate, propanoate and acetate (Supplementary Table S8, (11), (13), (15), (16)). C. proteolyticus strains could have been producing formate, butanol, butanoate and acetate (Supplementary Table S8, (1), (7), (11), (12)).
However, it is difficult to draw definite conclusions concerning the nature and number of the fermentation products because of incomplete information.
Syntrophic acetate oxidation and hydrogenotropic methanogenesis are the dominant pathways for methane production
At the metabolic level, most methanogens (including representatives of the Methanothermobacter genus) perform hydrogenotrophic methanogenesis exclusively; only members of the order Methanosarcinales perform acetoclastic methanogenesis or both methanogenesis pathways (Thauer et al., 2008). The data generated by pyrosequencing, metaproteomic approaches and isotopic analyses together strongly supported the production of methane through the hydrogenotrophic pathway by strains of the genus Methanothermobacter. Consistent with these results, thermophilic conditions are known to favour the hydrogenotrophic methanogenesis pathway (Schink, 1997; Hattori, 2008), but this is not systematically the case (for example, Hanreich et al., 2012). More precisely, among the 21 non-redundant protein groups identified as enzyme subunits required for hydrogenotrophic methanogenesis (Figure 3, Methanogens, steps 1–10 framed by a rectangle; Supplementary Table S9), three were specific for the hydrogenotrophic pathway, catalysing the reduction of CoM–S–S–CoB by H2 (Figure 3, Methanogens, hdrA, hdrC and mvhD genes, step 10, red). Moreover, none of the enzymes specific for acetoclastic methanogenesis were identified (Figure 3, Methanogens, steps 12, 13 and 15, blue). The identified proteins related to methanogenesis were mainly affiliated to Methanothermobacter members (Supplementary Table S9), consistent with a hydrogenotrophic pathway and with the pyrosequencing analyses (see above). Finally, the dominance of the hydrogenotrophic pathway was also supported by the apparent isotopic fractionation αC values determined over time (Figure 4, Supplementary Figure S3)(Conrad, 2005).

Identified enzymes involved in the methanogenesis and Acetyl-CoA pathways mapped over generic pathways. : proteins identified during the study. Red: hydrogenotrophic methanogenesis. Blue: acetoclastic methanogenesis. Green: methylotrophic methanogenesis. Purple: Eastern branch of the acetyl-CoA pathway. Orange: Western branch of the acetyl-CoA pathway. : membrane proteins. (Cofactors): key enzymes for the biosynthesis of cofactor. (1) Formylmethanofuran dehydrogenase, FwdA-F or FmdA-F; (2) Formylmethanofuran—tetrahydromethanopterin N-formyltransferase, Ftr; (3) Methenyltetrahydromethanopterin cyclohydrolase, Mch; (4) Methylenetetrahydromethanopterin dehydrogenase, Hmd/mtd; (5) Coenzyme F420-dependent N5,N10-methenyltetrahydromethanopterin reductase, Mer; (6) Tetrahydromethanopterin S-methyltransferase, Mtr; (7) Methyl-coenzyme M reductase, MrtABG or McrABG; (8) Membrane hydrogenase, Ech, Eha or Ehb, Rnf, Fpo; (9) Coenzyme F420 hydrogenase, Frh, Fru, Frc; (10) F420 non-reducing hydrogenase/heterodisulfide reductase complex, MvhADG-HdrABC or VhoACG-HdrDE; (11) ATPase, AhaA-K; (12) Acetate kinase, Ack; (13) Phosphotransacetylase, Pta; (14) Carbon monoxide dehydrogenase/acetyl-coA synthase complex, CODH/ACS; (15) Carbon monoxide dehydrogenase/acetyl-coA synthase/decarbonlyase (CODH/ACDS) complex; (16) Methylcobamide:CoM metyltransferase, MtaABC, Mtb, Mtm, Mtq, Mts or Mtt; (17) Formate dehydrogenase, Fdh; (18) Na+/H+ antiporter; (19) Coenzyme F390; (20) Formyltetrahydrofolate (CHO-THF) synthetase, Fhs or FTHFS; (21) Biofunctional protein—Methenyltetrahydrofolate (CH-THF) cyclohydrolase and methylenetetrahydrofolate (CH2-THF) dehydrogenase complex, FolD; (22) Methylenetetrahydrofolate (CH2-THF) reductase, MetF or MTHFR; (23) THF:Fe-S-Co Methyltransferase, MeTr. See also Supplementary Table S7.

Temporal evolution of the apparent fractionation factor αC calculated from the 13C isotopic signal of methane (δCH4) and CO2 (δCO2) in the biogas. For substrates of natural isotopic composition, such as office paper in the present case, αC values >1.065 are associated with hydrogenotrophic methanogenesis (Conrad, 2005). Measures were performed on the five replicates and on two controls. The mean values and s.d. are shown.
As acetate first accumulated during the degradation (Supplementary Figure S2) and was then consumed during the phase of biogas isotopic enrichment, it was reasonable to assume that acetate was transformed into H2 and CO2 by syntrophic acetate oxidation (SAO). The presence of syntrophic hydrogen suppliers in the community was therefore questioned (reviewed in Schink, 1997). Metaproteomic together with the pyrosequencing data brought some interesting insights, but definitive conclusions could not be drawn owing to the lack of known protein specific for the SAO pathways and to the still limited knowledge about thermophilic SAO microorganisms compared with mesophilic ones (reviewed in Hattori, 2008; Westerholm et al., 2011; and other publications from Schnürer’s group).
In the present case, the majority of the key bacterial enzymes involved in the Acetyl-CoA pathway were identified (Figure 3, Acetogens, steps 12–14), suggesting the presence of acetogenic bacteria. They represented eight non-redundant protein groups with an average of four peptides per group and 12% protein coverage (Supplementary Table S9). They included subunits from the carbon monoxide dehydrogenase/acetyl-coA synthase/decarbonlyase complex (Supplementary Table S9, step 14) assigned to Moorella thermoacetica (acsC gene), Carboxydothermus hydrogenoformans and Thermodesulfatator indicus (cooSI gene). Four formyltetrahydrofolate synthetase proteins (regarded as the key and specific enzymes for the oxidative Acetyl-CoA pathway, also named Eastern branch of the Wood-Ljungdahl pathway) were retrieved and assigned to Caldicellulosiruptor lactoaceticus (two of them), Desulfatibacillum alkenivorans and C. proteolyticus, respectively. Finally, a serine hydroxymethyltransferase was assigned to C. proteolyticus (Supplementary Table S9, step 22).
Comparing these results to the pyrosequencing data, C. proteolyticus and Gelria bacteria can be proposed as important H2 producers in the system and might have established efficient syntrophy with Methanothermobacter archaea (Plugge et al., 2002; Sasaki et al., 2011). However, according to the literature (Plugge et al., 2002; Sasaki et al., 2011) and to their protein expression profile, they corresponded to typical fermenting microorganisms rather than to obligate SAO. Based on the pyrosequencing data, other good candidates for SAO could be other members of the order Thermoanaerobacterales (Figure 2b). For instance, the family Thermoanaerobacteraceae comprises thermophilic and strictly anaerobic acetogenic species such as Thermoacetogenium phaeum (SAO, Hattori, 2008), M. thermoacetica (homoacetogen, Pierce et al., 2008) and C. hydrogenoformans (hydrogenogenic bacterium producing the carbon monoxide dehydrogenase/acetyl-coA synthase/decarbonlyase complex with the corresponding acs operon closely related to that of M. thermoacetica; Wu et al., 2005). The moderate proportion of pyrosequencing sequences attributed to the Thermoanaerobacterales order (∼0.4% without Gelria) seems similar to proportions observed for SAO bacteria in other systems (for example, Wersterholm et al., 2011). Strains belonging to the Clostridiales order may also have been performing SAO.
In conclusion, the microorganisms acting as SAO in syntrophy with the hydrogenotrophic methanogens could not be clearly identified, but the data indicated the presence of several strains closely related to known homoacetogens or SAO microorganisms. As presented above, the limited number of identified proteins related to SAO candidates was probably linked to the lack of closely related entries in the protein database.
Nitrogen metabolism and recycling: the abundance of proteolytic strains from the species C. proteolyticus
Among the numerous identified proteins putatively involved in ammonia assimilation and amino acid biosynthesis (Supplementary Table S10) and in peptidase activities (Supplementary Table S11), 22 were surprisingly affiliated to C. proteolyticus (11 in each of Supplementary Tables S10 and S11), indicating that C. proteolyticus strains could have been exerting an intensive proteolytic activity during the methanisation (Cai et al., 2011).
The extracellular protease activity from C. proteolyticus strains was first supported by the identification of a putative extracellular cell wall-attached protease (B5Y6Q5, MEROPS S8A). Based on MEROPS information (Rawlings et al., 2012), this protease could have a role in nutrition and it could have been abundantly produced because 13 peptides were detected by MS/MS analyses, the highest level among identified peptidases (Supplementary Table S10). In addition, a putative peptidase T from C. proteolyticus was possibly involved in general protein turnover (B5Y9V8, MEROPS M20). Very interestingly, three of the putative peptidases identified from C. proteolyticus were potentially involved in microcin synthesis (bacterial toxin composed of few peptides) (Duquesne et al., 2007; Cai et al., 2011). Indeed, they belonged to the three distinct UniRef50 clusters UniRef50_B5Y6N5 Tltd/PmbA, UniRef50_B5Y6N6 LmbIH and UniRef50_B5Y9Y2 TldD, and to the MEROPS family U62. This suggested that C. proteolyticus could have been actively predating other microorganisms in addition to simply scavenging extracellular proteinaceous material.
This proteolytic activity was further supported by other retrieved protein functions. In particular, seven ABC transporter clusters were attributed to C. proteolyticus including three related to peptide transport (B5Y898, B56YV2-B5Y6V3 and B5Y897), suggesting an important peptide import/export activity (Supplementary Table S12). Several proteins from C. proteolyticus related to oxidative stress response or to virulence factor were also identified, but their role remains unclear.
Finally, the important activity of C. proteolyticus members in the present anaerobic system was generally supported by the present body of data. One-hundred and four non-redundant protein groups were affiliated to C. proteolyticus (Figure 1c, Supplementary Table S18), among which 53 belonged to UniRef50 clusters specific to the species. About 30% of the 16S rRNA gene pyrotags were attributed to the species (Figure 2b), and a significant fraction of the bacterial community was hybridised with a probe targeting Corprothermobacter species (Supplementary Figure S11).
The proteolytic activity of Coprothermobacter members is clearly supported by the literature, and such strains were detected a number of times during anaerobic digestion of protein-rich waste (for example, Ollivier et al., 1985; Kersters et al., 1994; Etchebehere et al., 1998; Sasaki et al., 2007, 2011). It was furthermore reported that their ability to ferment carbohydrates was far inferior compared with that to ferment proteins (Ollivier et al., 1985; Kersters et al., 1994; Etchebehere et al., 1998). Their abundance in a system fed with paper as the sole exogenous substrate is surprising and suggests that extracellular proteinaceous material was abundant. Several distinct and non-exclusive protein sources can be suggested: the protein constituents of the EPS (up to 40% based on Liu and Fang, 2002); the abundant extracellular enzymes, in particular cellulosomes/cellulases or other proteins secreted by C. thermocellum (Ellis et al., 2012) and other cellulolytic strains; dead cell material present in the initial inoculum; and finally dead cell material generated during the incubation by the counter-selection of poorly competitive strains, by various stresses or by predation. Possible predators could be C. proteolyticus members, as discussed above, as well as viruses, whose presence is compatible with the four identified CRISPR-associated protein groups (Supplementary Table S15) (Bhaya et al., 2011).
Concluding remarks
Combining metaproteomic analyses and isotopic, chemical and molecular approaches, the present study provides one of the most comprehensive views of expressed biological functions during cellulosic waste methanisation, complementing the information gained from previous metagenomic or metatranscriptomic studies (for example, Krause et al., 2008; Schluter et al., 2008; Jaenicke et al., 2011; Rademacher et al., 2012; Zarkzewski et al., 2012). Using the complete UniprotKB database, over 500 protein functions were identified. The novel information gained on the microbial catalysts highlights the importance of ecological interactions between microbial groups, especially cooperation, for efficient methanisation. Based on the results, a model for the studied ecosystem is suggested (Figure 5). Its main features are as follows: the complementarity of distinct cellulolytic microbial groups to hydrolyse the recalcitrant substrate; the absence of acetoclastic methanogenesis despite the abundance of acetate as fermentation product; the dominance of hydrogenotrophic methanogenesis in association with syntrophic microorganisms; and finally the abundance of proteolytic fermentative Coprothermobacter strains. For a given microbial group, the identification level strongly relied on the availability of genome sequences from closely related strains. Consequently, the picture is still incomplete, and additional information could probably be gained concerning the function of Gelria and Tta-b61 with a more specific database that could typically be obtained by complementary metagenomic approaches.

Functional model for the anaerobic digestion of lignocellulose by complex thermophilic communities.
To our knowledge, the abundant proteolytic activity has never been reported for similar polysaccharide-fed bioprocesses. Further investigation of proteolytic activity in large-scale methanisation plants treating lignocellulosic waste could help to better understand the carbon and nitrogen fluxes during such processes. The probable complementarity of distinct cellulolytic strains is also an important aspect, and whether it is a general feature in similar systems remains an open question.
Metaproteomics appears as an attractive tool for providing direct and cost-limited access to functional information (reviewed in Wilmes and Bond, 2009; Schneider and Riedel, 2010). The exponential increase in sequence database size should reinforce the attractiveness of this approach in the future.
References
Abram F, Enright AM, O’Reilly J, Botting CH, Collins G, O’Flaherty V . (2011). A metaproteomic approach gives functional insights into anaerobic digestion. J Appl Microbiol 110: 1550–1560.
Bayer EA, Lamed R, White BA, Flint HJ . (2008). From cellulosomes to cellulosomics. Chem Rec 8: 364–377.
Beganovic J, Guillot A, van de Guchte M, Jouan A, Gitton C, Loux V et al (2010). Characterization of the insoluble proteome of Lactococcus lactis by SDS-PAGE LC-MS/MS leads to the identification of new markers of adaptation of the bacteria to the mouse digestive tract. J Proteome Res 9: 677–688.
Bhaya D, Davison M, Barrangou R . (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45: 273–297.
Cai H, Gu J, Wang Y . (2011). Protease complement of the thermophilic bacterium Coprothermobacter proteolyticus. In: Arabina HR, Tran Q-N (eds) Proceeding of the International Conference on Bioinformatics and Computational Biology BIOCOMP'11. ISBN: Las Vegas, Nevada, USA, pp 18–21.
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B . (2009). The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37: D233–D238.
Chen Y, Cheng JJ, Creamer KS . (2008). Inhibition of anaerobic digestion process: a review. Biores Technol 99: 4044–4064.
Chouari R, Le Paslier D, Daegelen P, Ginestet P, Weissenbach J, Sghir A . (2005). Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester. Environ Microbiol 7: 1104–1115.
Conrad R . (2005). Quantification of methanogenic pathways using stable carbon isotopic signatures: a review and a proposal. Org Geochem 36: 739–752.
Denef VJ, Shah MB, VerBerkmoes NC, Hettich RL, Banfield JF . (2007). Implications of strain- and species-level sequence divergence for community and isolate shotgun proteomic analysis. J Proteome Res 6: 3152–3161.
Duquesne S, Destoumieux-Garzon D, Peduzzi J, Rebuffat S . (2007). Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat Prod Rep 24: 708–734.
Ellis LD, Holwerda EK, Hogsett D, Rogers S, Shao X, Tschaplinski T et al (2012). Closing the carbon balance for fermentation by Clostridium thermocellum (ATCC 27405). Biores Technol 103: 293–299.
Etchebehere C, Pavan ME, Zorzopulos J, Soubes M, Muxi L . (1998). Coprothermobacter platensis sp. nov., a new anaerobic proteolytic thermophilic bacterium isolated from an anaerobic mesophilic sludge. Int J Syst Bacteriol 48: 1297–1304.
Ganidi N, Tyrrel S, Cartmell E . (2009). Anaerobic digestion foaming causes—a review. Biores Technol 100: 5546–5554.
Gold ND, Martin VJJ . (2007). Global view of the Clostridium thermocellum cellulosome revealed by quantitative proteomic analysis. J Bacteriol 189: 6787–6795.
Hamilton-Brehm SD, Mosher JJ, Vishnivetskaya T, Podar M, Carroll S, Allman S et al (2010). Caldicellulosiruptor obsidiansis sp. nov., an anaerobic, extremely thermophilic, cellulolytic bacterium isolated from Obsidian Pool, Yellowstone National Park. Appl Environ Microbiol 76: 1014–1020.
Hanreich A, Heyer R, Benndorf D, Rapp E, Pioch M, Reichl U et al (2012). Metaproteome analysis to determine the metabolically active part of a thermophilic microbial community producing biogas from agricultural biomass. Can J Microbiol 58: 917–922.
Hattori S . (2008). Syntrophic acetate-oxidizing microbes in methanogenic environments. Microbes Environ 23: 118–127.
Holm-Nielsen JB, Al Seadi T, Oleskowicz-Popiel P . (2009). The future of anaerobic digestion and biogas utilization. Biores Technol 100: 5478–5484.
Imachi H, Sekiguchi Y, Kamagata Y, Hanada S, Ohashi A, Harada H . (2002). Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate-oxidizing bacterium. Int J Syst Evol Microbiol 52: 1729–1735.
Jaenicke S, Ander C, Bekel T, Bisdorf R, Dröge M, Gartemann KH et al (2011). Comparative and joint analysis of two metagenomic datasets from a biogas fermenter obtained by 454-pyrosequencing. PLoS One 6: e14519.
Kersters I, Maestrojuan GM, Torck U, Vancanneyt M, Kersters K, Verstraete W . (1994). Isolation of Coprothermobacter proteolyticus from an anaerobic digest and further characterization of the species. Sys Appl Microbiol 17: 289–295.
Konstantinidis KT, Tiedje JM . (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 10: 504–509.
Krause L, Diaz NN, Edwards RA, Gartemann K-H, Krömeke H, Neuwger H et al (2008). Taxonomic composition and gene content of a methane producing microbial community isolated from a biogas reactor. J Biotechnol 136: 91–101.
Kröber M, Bekel T, Diaz NN, Goesmann A, Jaenicke S, Krause L et al (2009). Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J Biotechnol 142: 38–49.
Li T, Mazeas L, Sghir A, Leblon G, Bouchez T . (2009). Insights into networks of functional microbes catalysing methanization of cellulose under mesophilic conditions. Environ Microbiol 11: 889–904.
Liu H, Fang HH . (2002). Extraction of extracellular polymeric substances (EPS) of sludges. J Biotechnol 95: 249–256.
Lochner A, Giannone RJ, Keller M, Antranikian G, Graham DE, Hettich RL . (2011). Label-free quantitative proteomics for the extremely thermophilic bacterium Caldicellulosiruptor obsidiansis reveal distinct abundance patterns upon growth on cellobiose, crystalline cellulose, and switchgrass. J Proteome Res 10: 5302–5314.
Maki M, Leung KT, Qin W . (2009). The prospects of cellulase-producing bacteria for the bioconversion of lignocellulosic biomass. Int J Biol Sci 5: 500–516.
Ollivier BM, Mah RA, Ferguson TJ, Boone DR, Garcia JL, Robinson R . (1985). Emendation of the genus Thermobacteroides—Thermobacteroides-Proteolyticus sp-nov, a proteolytic acetogen from a methanogenic enrichment. Int J Syst Bacteriol 35: 425–428.
O’Sullivan C, Burrell PC, Clarke WP, Blackall LL . (2007). A survey of the relative abundance of specific groups of cellulose degrading bacteria in anaerobic environments using fluorescence in situ hybridization. J Appl Microbiol 103: 1332–1343.
Petersen TN, Brunak S, von Heijne G, Nielsen H . (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786.
Pierce E, Xie G, Barabote RD, Saunders E, Han CS, Detter JC et al (2008). The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ Microbiol 10: 2550–2573.
Plugge CM, Balk M, Zoetendal EG, Stams AJM . (2002). Gelria glutamica gen. nov., sp. nov., a thermophilic, obligately syntrophic, glutamate-degrading anaerobe. Int J Syst Evol Microbiol 52: 401–407.
Qu X, Mazéas L, Vavilin VA, Epissard J, Lemunier M, Mouchel J-M et al (2009). Combined monitoring of changes in delta13CH4 and archaeal community structure during mesophilic methanization of municipal solid waste. FEMS Microbiol Ecol 68: 236–245.
Rademacher A, Zakrzewski M, Schlüter A, Schönberg M, Szczepanowski R, Goesmann A et al (2012). Characterization of microbial biofilms in a thermophilic biogas system by high-throughput metagenome sequencing. FEMS Microbiol Ecol 79: 785–799.
Rawlings ND, Barrett AJ, Bateman A . (2012). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40: D343–D350.
Riviere D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J et al (2009). Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 3: 700–714.
Sasaki K, Haruta S, Ueno Y, Ishii M, Igarashi Y . (2007). Microbial population in the biomass adhering to supporting material in a packed-bed reactor degrading organic solid waste. Appl Microbiol Biotechnol 75: 941–952.
Sasaki K, Morita M, Sasaki D, Nagaoka J, Matsumoto N, Ohmura N et al (2011). Syntrophic degradation of proteinaceous materials by the thermophilic strains Coprothermobacter proteolyticus and Methanothermobacter thermautotrophicus. J Biosci Bioeng 112: 469–472.
Schink B . (1997). Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61: 262–280.
Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann KH et al (2008). The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol 136: 77–90.
Schneider T, Riedel K . (2010). Environmental proteomics: analysis of structure and function of microbial communities. Proteomics 10: 785–798.
Sokolova TG, Gonzalez JM, Kostrikina NA, Chernyh NA, Slepova TV, Bonch-Osmolovskaya EA et al (2004). Thermosinus carboxydivorans gen. nov., sp. nov., a new anaerobic, thermophilic, carbon-monoxide-oxidizing, hydrogenogenic bacterium from a hot pool of Yellowstone National Park. Int J Syst Evol Microbiol 54: 2353–2359.
Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R . (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Micro 6: 579–591.
van de Werken HJG, Verhaart MRA, VanFossen AL, Willquist K, Lewis DL, Nichols JD et al (2008). Hydrogenomics of the extremely thermophilic bacterium Caldicellulosiruptor saccharolyticus. Appl Environ Microbiol 74: 6720–6729.
van Lier JB, Tilche A, Ahring BK, Macarie H, Moletta R, Dohanyos M et al (2001). New perspectives in anaerobic digestion. Water Sci Technol 43: 1–18.
VanFossen AL, Verhaart MRA, Kengen SMW, Kelly RM . (2009). Carbohydrate utilization patterns for the extremely thermophilic bacterium Caldicellulosiruptor saccharolyticus reveal broad growth substrate preferences. Appl Environ Microbiol 75: 7718–7724.
Ward AJ, Hobbs PJ, Holliman PJ, Jones DL . (2008). Optimisation of the anaerobic digestion of agricultural resources. Biores Technol 99: 7928–7940.
Westerholm M, Dolfing J, Sherry A, Gray ND, Head IM, Schnürer A . (2011). Quantification of syntrophic acetate-oxidising microbial communities in biogas processes. Environ Microbiol Rep 3: 500–505.
Wilmes P, Bond PL . (2004). The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ Microbiol 6: 911–920.
Wilmes P, Bond PL . (2009). Microbial community proteomics: elucidating the catalysts and metabolic mechanisms that drive the Earth's biogeochemical cycles. Curr Opin Microbiol 12: 310–317.
Wu M, Ren Q, Durkin AS, Daugherty SC, Brinkac LM, Dodson RJ et al (2005). Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet 1: e65.
Yang SJ, Kataeva I, Hamilton-Brehm SD, Engle NL, Tschaplinski TJ, Doeppke C et al (2009). Efficient degradation of lignocellulosic plant biomass, without pretreatment, by the thermophilic anaerobe ‘Anaerocellum thermophilum’ DSM 6725. Appl Environ Microbiol 75: 4762–4769.
Zakrzewski M, Goesmann A, Jaenicke S, Jünemann S, Eikmeyer F, Szczepanowski R et al (2012). Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J Biotechnol 158: 248–258.
Zaneveld JR, Lozupone C, Gordon JI, Knight R . (2010). Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic Acids Res 38: 3869–3879.
Acknowledgements
Equipment at Irstea HBAN was acquired in the framework of the LABE project funded by CPER 2007–2013. The project was funded by Agence Nationale de la Recherche, project ANR Bioénergies/DANAC and by the project NSFC (21177096, 50878166). We acknowledge Elie Desmond for his valuable help and discussions on the taxonomic analyses and Yohan Rodolphe for his contribution to the sequencing experiments. The authors also would like to thank the anonymous reviewers for their helpful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Lü, F., Bize, A., Guillot, A. et al. Metaproteomics of cellulose methanisation under thermophilic conditions reveals a surprisingly high proteolytic activity. ISME J 8, 88–102 (2014). https://doi.org/10.1038/ismej.2013.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.120
Keywords
This article is cited by
-
Methane recovery from acidic tofu wastewater using an anaerobic fixed-bed reactor with bamboo as the biofilm carrier
Journal of Material Cycles and Waste Management (2021)
-
The Relationship Between the Gut Microbiome and Neurodegenerative Diseases
Neuroscience Bulletin (2021)
-
Soil metaproteomics as a tool for monitoring functional microbial communities: promises and challenges
Reviews in Environmental Science and Bio/Technology (2020)
-
Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants
Microbiome (2019)
-
From proteins to polysaccharides: lifestyle and genetic evolution of Coprothermobacter proteolyticus
The ISME Journal (2019)
