
Abstract
Terrestrial ecosystems are receiving elevated inputs of nitrogen (N) from anthropogenic sources and understanding how these increases in N availability affect soil microbial communities is critical for predicting the associated effects on belowground ecosystems. We used a suite of approaches to analyze the structure and functional characteristics of soil microbial communities from replicated plots in two long-term N fertilization experiments located in contrasting systems. Pyrosequencing-based analyses of 16S rRNA genes revealed no significant effects of N fertilization on bacterial diversity, but significant effects on community composition at both sites; copiotrophic taxa (including members of the Proteobacteria and Bacteroidetes phyla) typically increased in relative abundance in the high N plots, with oligotrophic taxa (mainly Acidobacteria) exhibiting the opposite pattern. Consistent with the phylogenetic shifts under N fertilization, shotgun metagenomic sequencing revealed increases in the relative abundances of genes associated with DNA/RNA replication, electron transport and protein metabolism, increases that could be resolved even with the shallow shotgun metagenomic sequencing conducted here (average of 75 000 reads per sample). We also observed shifts in the catabolic capabilities of the communities across the N gradients that were significantly correlated with the phylogenetic and metagenomic responses, indicating possible linkages between the structure and functioning of soil microbial communities. Overall, our results suggest that N fertilization may, directly or indirectly, induce a shift in the predominant microbial life-history strategies, favoring a more active, copiotrophic microbial community, a pattern that parallels the often observed replacement of K-selected with r-selected plant species with elevated N.
Similar content being viewed by others

Introduction
Ecosystems across the globe are receiving elevated levels of nitrogen (N): anthropogenic activities have doubled rates of N deposition and fixation compared with pre-industrial levels (Galloway et al., 2004). Rates of N deposition already exceed 10 kg N ha−1 yr−1 in regions of western Europe, southeast Asia and North America (Dentener et al., 2006), and the average global rate of N deposition to terrestrial ecosystems is likely to increase by a factor of 2.5 over the next century (Lamarque et al., 2005). Just as important, a large proportion of the global land surface is used for crop production where significant quantities of N (often >100 kg N ha−1 yr−1) are applied directly to soils as fertilizer.
As a key factor impacting terrestrial ecosystems worldwide, the effects of elevated N inputs on biota and biogeochemical cycles have been the focus of many studies. In most systems, elevating N inputs increases plant productivity (LeBauer and Treseder, 2008), with corresponding changes in plant community composition and, in most cases, decreases in local plant species diversity (Clark et al., 2007; Cleland and Harpole, 2010). It is also well-established that microbial taxa associated with specific components of the soil N cycle (for example, nitrifiers) often change in relative abundance when soils are amended with N. We know far less about how elevated N inputs will influence the structure, and associated functioning, of the entire belowground microbial community.
Recent studies have demonstrated N effects across a range of sites on both bacterial (for example, Campbell et al., 2010; Wessen et al., 2010) and fungal communities (for example, Frey et al., 2004; Egerton-Warburton et al., 2007). These shifts in the composition of soil microbial communities with N amendments are likely associated with shifts in the functional capabilities of these communities. Recent lab-based studies (for example, Strickland et al., 2009; Fukami et al., 2010) support this possibility by demonstrating that microbial communities are neither functionally redundant nor similar, with changes in their structure influencing rates of ecosystem-level processes. However, beyond the well-established direct effects of elevated N inputs on specific N cycling processes (Lu et al., 2011), it remains to be tested whether changes in the abundances of microbial taxa with N fertilization translate to changes in community functioning. To test this possibility, one promising approach is to pair phylogenetic analyses with shotgun metagenomics, the direct sequencing of whole-community DNA to characterize the genomic attributes of microbial communities. However, shotgun metagenomic analyses are rarely applied to replicated, experimental studies of soil microbial communities because it is implicitly assumed that deep sequencing is required for comparative analyses of highly diverse communities.
We used a suite of complementary approaches to determine how the structure and functional capabilities of soil microbial communities respond to long-term N amendments. We analyzed soils from two experimental N gradients that both received N fertilizer as NH4NO3, but have contrasting soil and vegetation types. The phylogenetic structure of the soil bacterial communities was determined by barcoded pyrosequencing (at least 1000 16S rRNA gene sequences per sample), and the functional potentials of the communities were characterized using catabolic assays and shotgun metagenomic profiling (with ≈75 000 sequences per sample). We used this unique data set to test the overarching hypothesis that prolonged increases in N availability shift the composition of soil microbial communities, their catabolic capabilities and their metagenomes. Such shifts may be, directly or indirectly, related to the changes in microbial carbon dynamics that are typically observed in soils receiving experimental N additions (Treseder, 2008; Janssens et al., 2010; Liu and Greaver, 2010), but the mechanisms underlying these changes in carbon dynamics remain largely undetermined.
Materials and methods
Sample collection
Soils were collected in July 2008 from two long-term N addition experiments: one located at the Cedar Creek Ecosystem Science Reserve (CC; Bethel, MN, USA) and one at the Kellogg Biological Station (KBS; Hickory Corners, MI, USA). The CC site was established in 1982 on successional grasslands. The KBS experiment was established in 2001 and is located in an agricultural field with a yearly rotating cropping system that is tilled at the end of each growing season. Both sites receive N fertilizer as NH4NO3, with the fertilizer applied either twice (CC) or once (KBS) per year. Detailed information on these two experiments is available online (http://www.cedarcreek.umn.edu and http://www.lter.kbs.msu.edu). Sample collection and processing methods have been described previously in Ramirez et al. (2010). For this study, we selected a subset of the soils included in that study for more detailed re-analysis. Specifically, we analyzed bulk soils from three N amendment levels per site (0, 34 and 272 kg N ha−1 yr−1 at CC; 0, 101 and 291 kg N ha−1 yr−1 at KBS), with soils collected from three replicate plots per N level (collecting and compositing six soil cores from randomly selected locations within each plot). Although no N fertilizer was applied to the control plots directly, these soils do receive N from atmospheric deposition (likely 10–40 kg N ha−1 yr−1, Dentener et al., 2006). Thus, we refer to these three N treatments as ‘low’, ‘medium’ and ‘high’ N levels. These levels were selected to represent current N deposition rates (the ‘low’ level), elevated atmospheric N deposition rates (the ‘medium’ level) and N amendment rates in intensively managed agricultural soils receiving direct fertilizer applications (the ‘high’ level). In total, 18 samples were included in this study (3 N levels × 2 sites × 3 replicate plots per N level per site). Characteristics of these sites and the collected soils are detailed in Supplementary Table S1, with soil properties and the effects of N on these properties discussed in detail in Ramirez et al. (2010). Plant biomass and composition at these sites have been described in Clark and Tilman (2008) and McSwiney and Robertson (2005).
Barcoded pyrosequencing of the 16S rRNA gene
We chose to focus solely on the composition and diversity of bacterial communities, because bacteria dominated the metagenomes in all 18 soils (see below) and archaea were rare in these soils (<3% of the 16S rRNA sequences; Bates et al., 2011). To analyze the composition of the bacterial communities, we used the procedure described in detail in Bates et al. (2011) and Lauber et al. (2009). Briefly, we used the MoBio Powersoil kit to extract DNA from 0.25 g of each of the 18 soil samples, with the soils carefully homogenized before subsampling. Amplifications were conducted in triplicate using the primer set 515f/806r that targets a region of the 16S rRNA gene that yields accurate phylogenetic information (Liu et al., 2007) and should have few biases against any bacterial taxa (Bates et al., 2011; Bergmann et al., 2011). The forward primer also included a 454 adapter, and the reverse primer contained both a 454 adapter and a 12-bp error-correcting barcode sequence. All amplicons were cleaned and pooled in equimolar concentrations into a single tube before sequencing on a Roche 454 GS FLX using the Titanium chemistry. The full length of the targeted 16S region was sequenced (approximately 300 bp reads, including the barcode) and, after quality filtering, all samples were rarefied to 1000 sequences per sample before conducting downstream analyses. All sequence analyses were conducted using the QIIME pipeline (Caporaso et al., 2010). Phylotypes were selected at the 97% sequence similarity level and the taxonomic identity was determined using the RDP scheme. Pairwise distances between communities were determined using unweighted Unifrac, a metric that measures the phylogenetic relatedness of whole communities and is well suited for comparing beta-diversity patterns between complex bacterial communities (Lozupone et al., 2006, 2011).
Shotgun metagenomic analyses
To obtain sufficient DNA from each of the 18 soil samples for metagenomic analyses (that is, analyses of whole community DNA via random sequencing, not just sequencing of specific, targeted genes), DNA was extracted from the soils (six replicate extractions per soil) using the MoBio PowerSoil kit as described above with modifications to the elution step. Instead of using separate aliquots of solution C6 to elute the DNA from each spin filter, the eluate collected from one spin filter served as the eluent for the next spin filter. Eluent collected from the first spin filter (100 μl) was pipetted onto the second filter and incubated again for 5 min at room temperature before centrifugation. These steps were carried out until all spin filters for a given sample had been eluted. Using this approach, DNA yields were between 1.0 and 2.5 μg per soil, as quantified using the PicoGreen dsDNA kit. The 18 DNA samples (1.0 μg per sample) were prepared for sequencing by nebulization, followed by tagging with GS-FLX-Titanium Rapid Library MID Adapters Kit (454 Life Sciences, Branford, CT, USA) and sequenced with the Titanium LR70 chemistry on a Roche 454 Gene Sequencer. The run yielded 518 Mbp of data, 1.35 million reads in total, with an average read length of 384 bp.
Sequences were assigned to subsystem categories using the MG-RAST web-server (Meyer et al., 2008), with a BLAST threshold of 1e-10 and a minimum read length of 50 bp. Only those reads that matched at least one subsystem category were included in downstream analyses. To estimate pairwise distances between metagenomes, the MG-RAST sequence assignments were first converted to percentages (% of assigned sequences per sample assigned to each category) with pairwise Euclidean distances calculated between each of the 18 samples.
Catabolic profiling
To compare the relative catabolic abilities of the microbial communities in each of the 18 soils, we used a catabolic profiling assay adapted from Degens and Vojvodic-Vukovic (1999). Briefly, 22 different organic carbon substrates (see list in Supplementary Table S4) were added individually to 4 g (dry wt. equivalent) of each soil. Net CO2 accumulation was measured on an infrared gas analyzer after an incubation period at 20 °C of 5 or 24 h (for the more recalcitrant substrates). The 22 substrates were chosen to represent a range of substrates potentially available to soil microorganisms, and included more recalcitrant substrates (chitin, cellulose, humic acid and lignin) as well as those that are more labile (amino acids, proteins, carboxylic acids and sugars). Substrates were added at concentrations ranging from 15 to 100 mM in 8 ml of solution with concentrations chosen following Degens and Vojvodic-Vukovic (1999). Assays were conducted in duplicate, yielding 828 assays in total (18 soils × 22 substrates plus the ‘water control’ × 2 analytical repeats per substrate per soil). Substrate catabolic rates were calculated as the mean difference from the ‘water controls’ (those samples that only received water) for each substrate per soil combination. As with the metagenomic data, pairwise distances in the catabolic potentials of the 18 soils were calculated using the Euclidean distance metric after expressing catabolic rates as percentages (CO2 production for a given substrate as a percentage of CO2 production for all substrates combined).
Statistical analyses
Relationships between samples were visualized using principal coordinate analyses (PCoA) from pairwise distances (Euclidean distances for the metagenomic and catabolic profiling data, unweighted Unifrac distances for the 16S data). Using the PRIMER software package (PRIMER-E, Plymouth, UK), the significance levels for differences between sample categories were determined using analysis of similarity tests, with correlations between pairwise distances from different assays determined using Mantel tests (Clarke and Gorley, 2006). Taxonomic and catabolic diversity levels per sample were estimated using the Shannon index (H′) as calculated from either phylotype relative abundances or percentages of CO2 production from each substrate.
Results and discussion
Composition of the bacterial communities across the N gradients
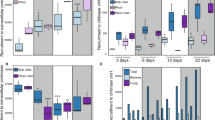
We analyzed soils from three N amendment levels per site, with soils collected from three replicate field plots per N level for a total of 18 samples. We obtained up to 3000 high-quality 16S rRNA gene sequences per sample, but all samples were rarefied to 1000 sequences per sample and thus one sample (CC23) was not included in any analyses of bacterial community structure due to insufficient sequence coverage. The soils from the Cedar Creek successional grassland site (CC) had slightly higher levels of bacterial taxonomic diversity (average H′=6.18, s.e.m.=0.04) than the agricultural site at the KBS (average H′=6.05, s.e.m.=0.03) (Figure 1), but there was no significant correlation between N addition level and phylotype diversity at either site (Spearman's r<0.2, P>0.2 in both cases; Figure 1). In contrast, Campbell et al. (2010) found a decrease in bacterial diversity with N additions, suggesting that the effects of N amendments on bacterial diversity levels are variable and likely site-dependent.

Comparison of taxonomic diversity (both bacteria and plants) and catabolic diversity for the plots studied at CC (a) and KBS (b). In all cases, diversity was calculated using Shannon's index (H′). KBS is a crop monoculture and thus plant species diversity is held constant across all nitrogen levels. Bars indicate 1 s.d.
The absence of any significant changes in bacterial diversity across the N gradient at CC contrasts with the plant community responses at this site, where plant diversity strongly decreased with increasing N additions (a pattern examined in detail in Clark and Tilman (2008) and references therein). This qualitative contrast between bacterial and plant diversity patterns across the N gradient suggests that the controls on plant diversity and bacterial diversity may not be the same and that one should not necessarily expect higher levels of bacterial diversity in soils with more plant species (Fierer and Jackson, 2006).
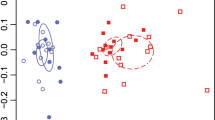
At both sites, the bacterial communities in the plots receiving the highest levels of N addition were significantly different from the communities receiving intermediate levels or no added N (Figure 2a). These high-N bacterial communities differed from those receiving lower amounts of N in having higher relative abundances of Proteobacteria (alpha-Proteobacteria at KBS and gamma-Proteobacteria at CC), Bacteroidetes and Actinobacteria, with lower relative abundances of Acidobacteria (Figure 3). These taxon-specific patterns were fairly consistent across the two sites, even though the sites are distinct with respect to site, soil and vegetation characteristics, a finding discussed in more detail in Ramirez et al. (2010). Bacteroidetes and many of the dominant actinobacterial and proteobacterial groups that increased in relative abundance across the gradient (Figure 3) have been putatively identified as being copiotrophic taxa (those taxa that thrive in conditions of elevated C availability and exhibit relatively rapid growth rates; Fierer et al., 2007; Eilers et al., 2010). In contrast, there was a decline in the abundance of Acidobacteria, a group that is often considered to be oligotrophic (Fierer et al., 2007; Davis et al., 2011) with slower growth rates and, in all likelihood, the ability to metabolize nutrient-poor and recalcitrant C substrates. Although these copiotrophic–oligotrophic categories are clearly over-simplifications of the broad range of ecological attributes and life-history strategies exhibited by soil microbes, previous work does allow us to identify bacterial taxa that likely fall into these general categories and our results are consistent with community-level copiotroph–oligotroph shifts across the N gradients. This presumed shift in the dominant life-history strategies may be a direct result of the increase in N availability as we would expect copiotrophic taxa to have lower biomass C:N ratios and higher N demands than more oligotrophic taxa (Fontaine and Barot, 2005; Fierer et al., 2007). Alternatively, the taxon-specific responses might be an indirect N effect arising from the increase in organic carbon availability associated with the increase in plant productivity in soils amended with N.

PCoA plots illustrating distances between microbial communities in individual samples. (a) Bacterial unweighted Unifrac distances (a measure of phylogenetic dissimilarity between communities). (b) Euclidean distances between metagenomes and (c) Euclidean distances between catabolic profiles. Letters within parentheses in the legends indicate groups of samples that were significantly different (P<0.01).

Changes in the relative abundances of bacterial taxa across the N gradients at the CC (a) and KBS sites (b). Only shown are those taxa with average relative abundances >1% at either of the two sites. The numbers on the right indicate the correlation (Spearman's ρ values) between N input rates and relative abundances calculated across all samples for that site. Bars indicate 1 s.d. For more details on the relative abundances of taxa in these soils, see Supplementary Table S2.
Similar taxon shifts to those we observed have been found in other studies examining bacterial community responses to N addition. For example, Campbell et al. (2010) noted that Acidobacteria were relatively less abundant and Proteobacteria (particularly gamma-Proteobacteria) were more abundant in fertilized plots, a pattern also observed in Wessen et al. (2010). This apparent consistency suggests that very high levels of N addition may have similar effects on the composition of bacterial communities across sites, but more comprehensive cross-site studies are required to test this hypothesis.
Characteristics of the metagenomes across the N gradients
Of the approximately 75 000 shotgun metagenomic sequences obtained per sample, only 25–35% of these sequences could be assigned to functional categories, percentages that are similar to those reported in other metagenomic surveys of environmental samples (Dinsdale et al., 2008; Mou et al., 2008). Although the metagenomic libraries were generated from bulk DNA extracted directly from the soil, the metagenomes are most likely reflective of the collective bacterial genomes as >98% of the ssu rRNA gene sequences identified from the metagenomic data were bacterial with, at most, 1–2 fungal ssu rRNA gene sequences detected compared to 70–130 bacterial ssu rRNA gene sequences per sample. Although the fungi can represent a significant proportion of belowground biomass (Strickland and Rousk, 2010), their minimal representation in the metagenome was likely a product of bacteria having higher rRNA gene concentrations per unit biomass.
The level of sequencing depth we used clearly resolved differences in metagenome composition; just as the soils from the two sites harbored distinct bacterial communities, they also differed in their metagenomic characteristics (Figure 2b). Also, within each site the characteristics of the metagenomes were significantly affected by N addition, with the soils receiving the highest levels of N having metagenomes that were significantly different from those receiving lower or no N inputs (Figure 2b). Although the metagenomes from the soils at the two sites were distinct, a subset of the specific gene categories that changed across the N gradients were similar between the two sites, suggesting that some of the soil metagenomic responses to N addition are similar across sites. The shifts in the relative abundances of specific gene categories observed across each of the N gradients (Figure 4) are consistent with the bacterial taxon responses, suggesting that the communities become more copiotrophic as N inputs increase. Indeed, we found that genes associated with DNA/RNA replication, electron transport and protein metabolism increased in relative abundance across the N gradients (Figure 4). We would expect all of these gene categories to be more common in copiotrophic than in oligotrophic bacteria (Fierer et al., 2007), but more detailed genomic analyses of bacteria from across the oligotroph–copiotroph continuum is required to confirm this hypothesis. At both sites, genes associated with urea decomposition are relatively less abundant in the plots receiving the highest levels of N, suggesting that the communities at both sites become less reliant on organic forms of N as additional fertilizer is applied. With the relatively shallow sequencing effort conducted here, many possible genes or gene categories were not sufficiently abundant in the data set to determine accurately changes in their relative abundances across the experiments. This includes genes associated with specific processes likely to be of critical importance to carbon and nitrogen cycling in these systems (including genes associated with N2 fixation, lignin degradation and ammonia oxidation). Deeper metagenomic sequencing will be required to capture these genes and to build a more comprehensive understanding of how the functional capabilities of the soil microbial communities are impacted by N fertilization.

Selected gene categories that changed in relative abundance across the N gradient, as estimated from the metagenomic data. For a full list of all gene categories and their relative abundances in each sample, see Supplementary Table S3. Data are expressed as z-scores, with red colors indicating higher z-scores (higher relative abundance compared with the mean for that gene category). (a, b) Results from the CC and KBS sites, respectively, with the header row indicating the specific plot numbers. Only those gene categories that were well correlated with nitrogen addition rates (ρ values >0.6 or <−0.6) are shown here. ρ Values for each gene category (relating relative gene abundances to N addition rates) are reported to the right of the heatmap along with the details on the mean percentage of assignable sequences in each gene category.
Catabolic diversity across the N gradients
The catabolic profiling assays provide a very different perspective on the functional capabilities of these communities across the N gradients as we measured the potential for these communities to catabolize a range of organic carbon substrates. As expected, all substrates were catabolized in each of the 18 soils examined. However, the soils were not equivalent in their relative catabolic capabilities. At CC, the soils receiving the highest levels of N had greater catabolic diversity than the soils receiving less or no added N inputs, a pattern that was reversed at KBS (Figure 1). Nitrogen addition level also clearly influenced the catabolic capabilities of the microbial communities (Figure 2c). Just as with the phylogenetic and metagenomic patterns (Figures2a and b, respectively), soils receiving the highest levels of N had significantly distinct catabolic capabilities from the unamended soils (Figure 2c), with minimal variability between replicate plots. However, the specific substrates that were favored at high N levels versus lower N levels were not the same at the two sites, that is, the shifts in the relative rates at which individual substrates were catabolized were not consistent across the two gradients (Figure 5). This may partly reflect phylogenetic or physiological responses of the microbial communities to differences in the amounts and/or types of organic C substrates available at each site across the N gradients (Degens et al., 2000; Frey et al., 2004). Alternatively, some of the catabolic shifts described in Figures 2c and 5 could be related to shifts in substrate use efficiencies or shifts in the fungal communities, which were poorly represented in the metagenomes, but could represent a significant portion of the active biomass pools (Strickland and Rousk, 2010). Advances in understanding links between microbial community structure and function in soils clearly relies on the development of methods to independently assess the functioning of bacterial and fungal assemblages in intact microbial communities or molecular tools that permit simultaneous characterization of the whole community at resolutions proportional to bacterial and fungal activities.

Selected substrates for which the relative catabolic rates changes across the N gradients, as estimated from the catabolic profiling data. For a full list of all substrates and their relative catabolism in each sample, see Supplementary Table S4. Data are expressed as z-scores, with red colors indicating higher z-scores (higher relative abundance compared with the mean for that gene category). (a, b) Results from the CC and KBS sites, respectively, with the header row indicating the specific plot numbers. Only those substrates with catabolic rates that were well correlated with N addition rates (ρ values >0.6 or <−0.6) are shown here. ρ Values for each substrate (relating catabolic rates to N addition rates) are reported to the right of the heatmap along with the mean relative catabolic rate (expressed as a percentage of the catabolic rate for all substrates combined for each sample).
Methodological considerations
As demonstrated in this and other studies, metagenomics provides a powerful tool for studying the functional capacities of individual microbial communities. However, the wider application of metagenomic tools to compare microbial community attributes requires sufficient statistical replication to confirm the validity of any observed patterns. Without replicated metagenomic data, many of the observed differences in metagenome characteristics across sample categories or experimental treatments are merely anecdotal (Prosser, 2010). Field replication is missing from nearly all published metagenomic studies owing, in part, to the assumption that deep sequencing of complex microbial communities is required for robust comparisons of metagenomes. Here we show that this is not the case. Even at a relatively shallow sequencing depth of <100 000 reads per sample, we were able to differentiate the metagenomes across sites and experimental treatments, identifying key gene categories that change in relative abundance across the N gradients. For studies where there is interest in genes that are relatively rare (for example, studies looking for specific genes involved in ammonia oxidation or lignin breakdown) or interest in the detailed phylogenetic structure of the communities, the metagenomic approach used here would be nearly useless as far deeper sequencing would be required to document changes in the relative abundances of relatively rare genes or taxa across samples. However, as has been demonstrated previously for 16S rRNA-based surveys (Lauber et al., 2009), conducting shallower metagenomic analyses on more samples allows us to detect community-level ecological patterns and confirm their significance. Indeed, our data suggest that advances in the understanding of whole microbial communities, especially with regards to broadly dispersed physiological functions (such as those involved in general C metabolism), are feasible with relatively shallow and replicated metagenomic assessments.
Concordance between phylogenetic, metagenomic and functional changes in the communities
All three assays showed significant changes in microbial community characteristics across both the KBS and CC N gradients (Figure 2). Interestingly, the phylogenetic, metagenomic and catabolic responses were significantly correlated with one another in nearly all cases (Table 1), suggesting some degree of correspondence between these very different metrics of microbial community characteristics in how they are impacted by N amendments. However, it is important to note that the phylogenetic and metagenomic shifts were most pronounced at the highest N levels, with the intermediate N addition levels at both sites yielding communities that were not significantly different from the control plots. This observation is surprising because even the intermediate N levels (34 and 101 kg N ha−1 yr−1 at CC and KBS, respectively) are higher than most soils likely receive from chronic N deposition in all but the most heavily polluted regions (Dentener et al., 2006). In addition, these intermediate levels of N did lead to pronounced increases in plant productivity at both sites and changes in plant community composition at CC (McSwiney and Robertson, 2005; Clark and Tilman, 2008), suggesting that, in general, plant communities may be more sensitive to N additions than soil bacterial communities. It is possible that the direct effects of N fertilization on soil bacterial communities are subtle in all but those soils (like agricultural soils) that receive large, direct inputs of N fertilizer. The meta-analysis of field and laboratory N amendment studies conducted by Treseder, 2008 supports this hypothesis, as the largest effects of N addition on microbial biomass and activity were observed at the highest fertilization rates.
Long-term environmental manipulations cannot be used to tease out causation from correlation in microbial composition and functioning. However, such manipulations do provide observations that inform the development and testing of theory and, in this regard, the apparent predictability of the relationships between the phylogenetic and functional community properties found here is intriguing. Indeed, we found that phylogeny is correlated with metagenomic characteristics, suggesting that both phylogenetic and metagenomic information could be used to predict relative shifts in the catabolic characteristics of soil communities. These results add to the growing body of evidence that accurate prediction of how some ecosystem processes will respond to disturbance requires the explicit incorporation of soil microbes into ecosystem models (Allison and Martiny, 2008). That is, soil microbial communities cannot be assumed to be functionally redundant nor similar. Future work is required to investigate whether relationships between microbial community phylogeny, metagenome and catabolic functioning are generally observed at local scales in response to environmental perturbation and whether these shifts influence ecosystem processes in a manner not predictable from abiotic environmental parameters alone.
As evident from cross-biome (Dinsdale et al., 2008) and cross-taxon (Muegge et al., 2011) studies, shifts in the relative abundances of specific lineages generally lead to corresponding shifts in metagenome characteristics. We show that this same pattern holds at local scales in response to environmental perturbation. Our data are consistent with observations that major bacterial groups show coherent ecological and genomic characteristics (Philippot et al., 2010), and the often strong agreement between 16S rRNA distance and gene content conservation (Konstantinidis and Tiedje, 2005) or similarity in functional pathways (Chaffron et al., 2010). There will obviously be many gene categories for which shifts in relative abundances are not predictable from phylogenetic information (for example, those genes readily transferred horizontally) and we lack basic information on the genomic properties of many soil taxa. However, because metagenomic analyses are not trivial, future studies clearly have to ascertain whether (or in what circumstances) metagenomic sequencing provides ‘added value’, or if metagenomic shifts could largely be predicted from phylogenetic information. For example, large deviations of measured metagenomes from the baseline prediction generated by 16S rRNA/reference genome analysis (after appropriate corrections for genome size and phylogenetic distance; Konstantinidis and Tiedje, 2005; Chaffron et al., 2010) might imply ecologically important features of local habitat adaptation.
Possible mechanisms underlying the observed patterns
Both the phylogenetic and metagenomic data suggest that the N additions may result in a shift from a more oligotrophic bacterial community to one that is more copiotrophic, just as fertilizer often increases the abundance of r-selected ‘weedy’ plant species over longer-lived, slower-growing K-selected plant species (Suding et al., 2005; Cleland and Harpole, 2010). This overall shift in life-history strategies, with copiotrophic microbes increasing in relative abundance in heavily fertilized soils, may explain the widely observed decrease in soil microbial respiration rates and biomass with added N (Treseder, 2008; Janssens et al., 2010; Liu and Greaver, 2010), even though N additions typically increase both plant productivity and the quality of plant litter inputs to soil (Chapin et al., 1986; Suding et al., 2005). Copiotrophic taxa may be expected to have higher rates of activity per unit biomass, higher turnover rates and higher substrate use efficiencies yielding a smaller standing microbial biomass pool with faster growth rates. However, copiotrophic taxa should also be less likely to access more recalcitrant pools of C (Fontaine et al., 2004; Fierer et al., 2007; Miki et al., 2010), reducing overall rates of heterotrophic respiration as the communities preferentially consume more labile substrates over the more recalcitrant organic C substrates that make up the bulk of the soil organic C pool (akin to eating dessert and ignoring the other meal courses) (Fontaine and Barot, 2005). This oligotroph–copiotroph switch may complement other hypotheses posed to explain the inhibition of microbial activities with N fertilization, namely the N mining hypothesis (Craine et al., 2007) and the extracellular inhibition hypothesis (Fog, 1988; Sinsabaugh, 2010). Of course, the validity of our hypothesis remains undetermined and further studies are required to determine if this presumed oligotroph–copiotroph switch is a more universal response to N additions and if the hypothesis does indeed explain the soil microbial responses to N amendments.
References
Allison SD, Martiny JBH . (2008). Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci USA 105: 11512–11519.
Bates S, Caporaso JG, Walters WA, Knight R, Fierer N . (2011). A global-scale survey of archaeal abundance and diversity in soils. ISME J 5: 908–917.
Bergmann G, Bates S, Eilers K, Lauber C, Caporaso J, Walters W et al. (2011). The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43: 1450–1455.
Campbell BJ, Polson SW, Hanson TE, Mack MC, Schuur EAG . (2010). The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ Microbiol 12: 1842–1854.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7: 335–336.
Chaffron S, Rehrauer H, Pernthaler J, von Mering C . (2010). A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res 20: 947–959.
Chapin FS, Vitousek PM, VanCleve K . (1986). The nature of nutrient limitation in plant communities. Am Nat 127: 48–58.
Clark CM, Cleland EE, Collins SL, Fargione JE, Gough L, Gross KL et al. (2007). Environmental and plant community determinants of species loss following nitrogen enrichment. Ecol Lett 10: 596–607.
Clark CM, Tilman D . (2008). Loss of plant species after chronic low-level nitrogen deposition to prairie grasslands. Nature 451: 712–715.
Clarke K, Gorley R . (2006). PRIMER, 6th edn. PRIMER-E Ltd: Plymouth, UK.
Cleland EE, Harpole WS . (2010). Nitrogen enrichment and plant communities. Annals New York Acad Sci 1195: 46–61.
Craine JM, Morrow C, Fierer N . (2007). Microbial nitrogen limitation increases decomposition. Ecology 88: 2105–2113.
Davis KER, Sangwan P, Janssen PH . (2011). Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ Microbiol 13: 798–805.
Degens BP, Schipper LA, Sparling GP, Vojvodic-Vukovic M . (2000). Decreases in organic C reserves in soils can reduce the catabolic diversity of soil microbial communities. Soil Biol Biochem 32: 189–196.
Degens BP, Vojvodic-Vukovic M . (1999). A sampling strategy to assess the effects of land use on microbial functional diversity in soils. Aus J Soil Res 37: 593–601.
Dentener F, Drevet J, Lamarque JF, Bey I, Eickhout B, Fiore AM et al. (2006). Nitrogen and sulfur deposition on regional and global scales: a multimodel evaluation. Global Biogeochem Cycle 20: GB4003.
Dinsdale EA, Edwards RA, Hall D, Angly F, Breitbart M, Brulc JM et al. (2008). Functional metagenomic profiling of nine biomes. Nature 452: 629–632.
Egerton-Warburton LM, Johnson NC, Allen EB . (2007). Mycorrhizal community dynamics following nitrogen fertilization: a cross-site test in five grasslands. Ecol Monogr 77: 527–544.
Eilers KG, Lauber CL, Knight R, Fierer N . (2010). Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Biol Biochem 42: 896–903.
Fierer N, Bradford M, Jackson R . (2007). Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364.
Fierer N, Jackson R . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Fog K . (1988). The effect of added nitrogen on the rate of decomposition of organic matter. Biol Rev 63: 433–462.
Fontaine S, Bardoux G, Abbadie L, Mariotti A . (2004). Carbon input to soil may decrease soil carbon content. Ecol Lett 7: 314–320.
Fontaine S, Barot S . (2005). Size and functional diversity of microbe populations control plant persistence and long-term soil carbon accumulation. Ecol Lett 8: 1075–1087.
Frey SD, Knorr M, Parrent JL, Simpson RT . (2004). Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. Forest Ecol Man 196: 159–171.
Fukami T, Dickie IA, Wilkie JP, Paulus BC, Park D, Roberts A et al. (2010). Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol Lett 13: 675–684.
Galloway JN, Dentener FJ, Capone DG, Boyer EW, Howarth RW, Seitzinger SP et al. (2004). Nitrogen cycles: past, present, and future. Biogeochemistry 70: 153–226.
Janssens IA, Dieleman W, Luyssaert S, Subke JA, Reichstein M, Ceulemans R et al. (2010). Reduction of forest soil respiration in response to nitrogen deposition. Nat Geosci 3: 315–322.
Konstantinidis KT, Tiedje JM . (2005). Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102: 2567–2572.
Lamarque JF, Kiehl JT, Brasseur GP, Butler T, Cameron-Smith P, Collins WD et al. (2005). Assessing future nitrogen deposition and carbon cycle feedback using a multimodel approach: analysis of nitrogen deposition. JGR-Atmos 110; doi:10.1029/2005JD005825.
Lauber C, Knight R, Hamady M, Fierer N . (2009). Soil pH as a predictor of soil bacterial community structure at the continental scale: a pyrosequencing-based assessment. Appl Environ Microbiol 75: 5111–5120.
LeBauer DS, Treseder KK . (2008). Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology 89: 371–379.
Liu LL, Greaver TL . (2010). A global perspective on belowground carbon dynamics under nitrogen enrichment. Ecol Lett 13: 819–828.
Liu Z, Lozupone C, Hamady M, Bushman F, Knight R . (2007). Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35: e120.
Lozupone C, Hamady M, Knight R . (2006). UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinf 7: 371.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R . (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J 5: 169–172.
Lu M, Yang Y, Luo Y, Fang C, Zhou X, Chen J et al. (2011). Responses of ecosystem nitrogen cycle to nitrogen addition: a meta-analysis. New Phytol 189: 1040–1050.
McSwiney CP, Robertson GP . (2005). Nonlinear response of N2O flux to incremental fertilizer addition in a continuous maize (Zea mays L.) cropping system. Global Change Biol 11: 1712–1719.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M et al. (2008). The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf 9: 386.
Miki T, Ushio M, Fukui S, Kondoh M . (2010). Functional diversity of microbial decomposers facilitates plant coexistence in a plant–microbe–soil feedback model. Proc Natl Acad Sci USA 107: 14251–14256.
Mou XZ, Sun SL, Edwards RA, Hodson RE, Moran MA . (2008). Bacterial carbon processing by generalist species in the coastal ocean. Nature 451: 708–711.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332: 970–974.
Philippot L, Andersson SGE, Battin TJ, Prosser JI, Schimel JP, Whitman WB et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat Rev Microbiol 8: 523–529.
Prosser JI . (2010). Replicate or lie. Environ Microbiol 12: 1806–1810.
Ramirez K, Lauber C, Knight R, Bradford M, Fierer N . (2010). Consistent effects of nitrogen fertilization on the phylogenetic composition of soil bacterial communities in contrasting systems. Ecology 91: 3463–3470.
Sinsabaugh RL . (2010). Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biol Biochem 42: 391–404.
Strickland M, Lauber C, Fierer N, Bradford M . (2009). Testing the functional significance of microbial community composition. Ecology 90: 441–451.
Strickland MS, Rousk J . (2010). Considering fungal:bacterial dominance in soils—methods, controls, and ecosystem implications. Soil Biol Biochem 42: 1385–1395.
Suding KN, Collins SL, Gough L, Clark C, Cleland EE, Gross KL et al. (2005). Functional- and abundance-based mechanisms explain diversity loss due to N fertilization. Proc Natl Acad Sci USA 102: 4387–4392.
Treseder KK . (2008). Nitrogen additions and microbial biomass: a meta-analysis of ecosystem studies. Ecol Lett 11: 1111–1120.
Wessen E, Hallin S, Philippot L . (2010). Differential responses of bacterial and archaeal groups at high taxonomical ranks to soil management. Soil Biol Biochem 42: 1759–1765.
Acknowledgements
We thank members of the Fierer lab and three anonymous reviewers for valuable comments on previous drafts of this manuscript and Joe Jones at Engencore for his help with the 454 sequencing. We thank the members of the CC LTER, including David Tilman and Linda Kinkel, and the members of KBS LTER, particularly Jay Lennon and Zach Aanderud, for assisting with sample collection. Funding for this work was provided by the Howard Hughes Medical Institute (RK) the National Institutes of Health (RK), the US Department of Agriculture (NF), the National Science Foundation (NF, RK), the Andrew W Mellon Foundation (NF, MAB) and the US Department of Energy (MAB).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Fierer, N., Lauber, C., Ramirez, K. et al. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J 6, 1007–1017 (2012). https://doi.org/10.1038/ismej.2011.159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.159
Keywords
This article is cited by
-
Land management shapes drought responses of dominant soil microbial taxa across grasslands
Nature Communications (2024)
-
Effect of nitrogen reduction by chemical fertilization with green manure (Vicia sativa L.) on soil microbial community, nitrogen metabolism and and yield of Uncaria rhynchophylla by metagenomics
Archives of Microbiology (2024)
-
High nitrogen fertilizer input enhanced the microbial network complexity in the paddy soil
Soil Ecology Letters (2024)
-
Legacy effects of preceding crops improve flue-cured tobacco productivity in southwest China by optimizing soil structure, nutrients, and microbial interactions
Plant and Soil (2024)
-
Changes in Microbial Composition During the Succession of Biological Soil Crusts in Alpine Hulun Buir Sandy Land, China
Microbial Ecology (2024)
