
Abstract
To investigate the effects of mesoscale eddies on prokaryotic assemblage structure and activity, we sampled two cyclonic eddies (CEs) and two anticyclonic eddies (AEs) in the permanent eddy-field downstream the Canary Islands. The eddy stations were compared with two far-field (FF) stations located also in the Canary Current, but outside the influence of the eddy field. The distribution of prokaryotic abundance (PA), bulk prokaryotic heterotrophic activity (PHA), various indicators of single-cell activity (such as nucleic acid content, proportion of live cells, and fraction of cells actively incorporating leucine), as well as bacterial and archaeal community structure were determined from the surface to 2000 m depth. In the upper epipelagic layer (0–200 m), the effect of eddies on the prokaryotic community was more apparent, as indicated by the higher PA, PHA, fraction of living cells, and percentage of active cells incorporating leucine within eddies than at FF stations. Prokaryotic community composition differed also between eddy and FF stations in the epipelagic layer. In the mesopelagic layer (200–1000 m), there were also significant differences in PA and PHA between eddy and FF stations, although in general, there were no clear differences in community composition or single-cell activity. The effects on prokaryotic activity and community structure were stronger in AE than CE, decreasing with depth in both types of eddies. Overall, both types of eddies show distinct community compositions (as compared with FF in the epipelagic), and represent oceanic ‘hotspots’ of prokaryotic activity (in the epi- and mesopelagic realms).
Similar content being viewed by others

Introduction
Mesoscale eddies are ubiquitous features in the ocean (Cheney and Richardson, 1976; Arístegui et al., 1997; van Haren et al., 2006), with strong implications on regional biogeochemistry and productivity. Anticyclonic eddies (AEs) have been seen to accumulate organic matter within their cores (for example Arístegui et al., 2003; Mathis et al., 2007) and to exhibit elevated microbial respiration (Arístegui and Montero, 2005; Mouriño-Carballido and McGillicuddy, 2006) and heterotrophic production (Baltar et al., 2007; Ewart et al., 2008). Cyclonic eddies (CEs) are known to enhance nutrient inputs to the surface ocean increasing new production (Falkowsky et al., 1991; Harris et al., 1997; Morán et al., 2001) and chlorophyll concentrations (Arístegui et al., 1997; McGillicuddy Jr et al., 1998; Tarran et al., 2001). Current estimates suggest that up to 50% of the global new primary production may be caused by eddy-induced nutrient fluxes (Falkowsky et al., 1991; McGillicuddy Jr et al., 1998; Letelier et al., 2000). Thus, eddies exert a major control on the generation, accumulation, and downward transport of biogenic production in the ocean, as well as on the associated remineralization processes mediated by prokaryotes.
Despite the recognized important function of prokaryotes within the marine biogeochemical cycles (for example Azam et al., 1983), only a reduced number of studies, sometimes contradictory, have been published analyzing the response of heterotrophic prokaryotes to eddy activity. Some of these studies reported increased prokaryotic abundance (PA) inside cold-core eddies (Lochte and Pfannkuche, 1987; Harris et al., 1997; Thyssen et al., 2005) and in the frontal waters between CEs and AEs (Arístegui and Montero, 2005). Other studies, however, did not find differences in depth-integrated prokaryotic biomass between the inside and outside of CEs (González et al., 2001; Tarran et al., 2001). In the Canary Islands region, higher prokaryotic heterotrophic production rates were measured within eddies compared with the surrounding waters (Bode et al., 2001; Baltar et al., 2007). In the Sargasso Sea, Ewart et al. (2008) found an increase in prokaryotic heterotrophic production at the periphery of a CE relative to the eddy center, as well as in the core of an AE. The latter authors found a tight coupling between phytoplankton and prokaryotic activity, suggesting that the variability of phytoplankton community structure has an important function influencing prokaryotic heterotrophic production in these mesoscale features.
Less information is available concerning changes in prokaryotic community structure because of eddy influence. In a DMSP-producing coccolithophorid bloom in a North Atlantic cold-core eddy, González et al. (2000) found that Roseobacter, SAR86, and SAR11 were the dominant groups of Bacteria associated with the bloom. However, no differences in the dominant groups were found between inside and outside the eddy. In contrast, Benitez-Nelson et al. (2007) reported mixed-layer bacterioplankton communities being similar inside and outside a CE, but below 50 m depth Planctomycetes, Bacteroidetes, and certain Proteobacteria (thought to degrade high-molecular weight dissolved organic matter) were present. Zhang et al. (2009) found a greater crenarcheaal contribution in the upper mesopelagic waters inside two CEs (as compared with outside) that they related to a higher contribution of refractory dissolved organic matter. They also found a significantly higher bulk D-:L-Aspartic acid uptake ratio in the core of two CEs as compared with the outside areas, but no influence of the CEs was found in the ratio of D-:L-Aspartic acid positive cells of Bacteria and Archaea. However, information on Archaea and on the activity of prokaryotes at the single-cell level comparing cyclonic and anticyclonic features is not available.
Here, we report the abundance, relative nucleic acid content, viability, bulk and single-cell activities, and community structure of prokaryotic assemblages in four island-induced eddies (two CE and two AE), compared with two unaffected (far-field [FF]) reference sites northwest of the Canary archipelago. We examined the effect of eddies in an oligotrophic region, in which the impact on the prokaryotic community should be significant. On the basis of earlier studies on the function of mesoscale eddies in oceanic biogeochemistry and productivity, we hypothesize that eddies could generate oceanic ‘hotspots’ of activity and shifts in prokaryote assemblage composition at least in the epipelagic (0–200 m) and mesopelagic (200–1000 m) layers. We also investigated whether bathypelagic (1000–4000 m depth) prokaryotic assemblages underneath eddies respond to the presumably elevated vertical carbon fluxes.
Materials and methods
Study site and sampling
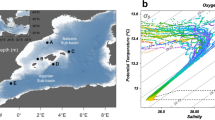
The positions of mesoscale eddies were deduced at first instance by satellite images of sea-surface temperature during cruise RODA-I (11 August to 9 September 2006) on board the RV ‘Hespérides’ (Figure 1). Once at the supposed eddy sites, their structures were characterized by means of XBT (expendable bathythermographs). Once located, the eddy center, temperature, salinity, and fluorescence were recorded down to 2000 m depth using a SeaBird 911 plus CTD system, mounted on a General Oceanics rosette sampler, equipped with 24 12 l Niskin bottles. Eddy stations were compared with two FF stations situated northwest of the Canary archipelago. These stations were placed inside the Canary Current, but outside the influence of the eddy field. Samples for PA and heterotrophic activity and nucleic acid content were collected at each station from 10 to 13 depths ranging from 5 to 2000 m, including the deep chlorophyll maximum (40–125 m), the deep scattering layer (450–550 m), and the oxygen minimum zone (720–850 m).

Position of the sampled stations located in the far-field (FF) and in the core of cyclonic eddy (CE) and anticyclonic eddy (AE) in the Canary Current system, during the RODA I cruise in August 2007. Sections crossing CE1 and AE1 are represented in Figure 3.
PA, nucleic acid content, and membrane-compromised bacteria determined by flow cytometry
Picoplankton collected from the different depth layers of the water column were enumerated using flow cytometry with an FACSCalibur (Becton-Dickinson, Franklin Lakes, NJ, USA) with a laser emitting at 488 nm. Samples (1.5 ml) were fixed with paraformaldehyde (1% final concentration), incubated at 4 °C for 15–30 min, and then stored frozen in liquid nitrogen until analysis. Before counting the cells by flow cytometry and after unfreezing, 200 μl of sample were stained with a DMSO-diluted SYTO-13 (Molecular Probes, Eugene, OR, USA) stock (10:1) at 2.5 μM final concentration. Prokaryotes were identified by their signatures in a plot of side scatter (SSC) versus green fluorescence (FL1). High nucleic acid (HNA) and low nucleic acid cells were separated in the scatter plot of SSC–FL1 (Gasol et al., 1999). HNA cells exhibited higher FL1 than low nucleic acid cells. Picocyanobacteria were discriminated in a plot of FL1 versus red fluorescence (FL3).
Viable and damaged prokaryotic cells were estimated in non-fixed samples after the nucleic acid double-staining (NADS) protocol (Gregori et al., 2001; Falcioni et al., 2008). NADS+, green cells (assumed to be active, with intact membranes), and NADS−, red cells (assumed to be inactive, with compromised cell membranes) were identified by simultaneous double staining with a membrane-permeant (SYBR Green; Molecular Probes) and impermeant (propidium iodide) probe. Immediately after collecting the samples, they were incubated in the dark with the probes for 15 min. NADS+ and NADS− cells were enumerated by flow cytometry and differentiated in a scatter plot of FL1 (green) -FL3 (red emission after blue-light excitation). Samples for PA and NADS were run at a flow rate of ∼60–70 μl min−1, which was determined volumetrically after every 10 samples run.
Prokaryotic heterotrophic activity estimated by [3H] leucine incorporation
Prokaryotic heterotrophic activity (PHA) was estimated from the incorporation of tritiated leucine using the centrifugation method (Smith and Azam, 1992). 3H-Leucine (Leu; Amersham, Little Chalfont, UK; specific activity=171 Ci mmol−1) was added at saturating concentration (40 nmol l−1) to 4 replicate 1.2 ml subsamples. Duplicate controls were established by adding 120 μl of 50% trichloroacetic acid 10 min before isotope addition. The Eppendorf tubes were incubated at in situ temperature in temperature-controlled chambers for 2–7 h. Incorporation of leucine in the quadruplicate sample was stopped by adding 120 μl ice-cold 50% trichloroacetic acid. Subsequently, the subsamples and the controls were kept at −20 °C until centrifugation (at ca. 12 000 g) for 20 min, followed by aspiration of the water. Finally, 1 ml of scintillation cocktail was added to the Eppendorf tubes before determining the incorporated radioactivity after 24–48 h on a Wallac scintillation counter with quenching correction using an external standard.
Catalyzed reporter deposition-fluorescence in situ hybridization
Immediately after collecting the samples from the Niskin bottles, 10–40 ml subsamples were fixed with paraformaldehyde (2% final concentration) and stored at 4 °C in the dark for 12–18 h. The cells were collected on 0.2 μm polycarbonate filters (Millipore, Bedford, MA, USA; GTTP, 25 mm filter diameter) supported by cellulose nitrate filters (Millipore, HAWP, 0.45 μm), washed twice with 0.2 μm-filtered Milli-Q water, dried, and stored in a microfuge vial at −20 °C until further processing in the laboratory. The filters were embedded in low-gelling-point agarose and incubated either with lysozyme for the Bacteria probes Eub338-III (mixture of probes Eub338, Eub II, and Eub III; Amann et al., 1990; Daims et al., 1999), or Proteinase-K for the marine Euryarchaeota Group II probe Eury806 and for the marine Crenarchaeota Group I probe Cren537 (Teira et al., 2004) and GI-554 (Massana et al., 1997). To determine the coverage of the two Crenarchaeota probes, hybridization was performed on a set of samples with the oligonucleotide probes Cren 537 and GI-554 separately, and applied as a mix (CrenTotal). Filters were cut in sections and hybridized with horseradish peroxidase-labeled oligonucleotide probes and tyramide-Alexa488 for signal amplification, after the protocol described in Teira et al. (2004). Cells were counter-stained with a DAPI mix: 5.5 parts Citifluor, 1 part Vectashield (Vector Laboratories, Burlingame, CA, USA), and 0.5 parts phosphate-buffered saline with DAPI (final concentration 1 μg ml−1). The slides were examined under a Zeiss Axioplan 2 microscope equipped with a 100 W Hg lamp and appropriate filter sets for DAPI and Alexa488. More than 800 DAPI-stained cells were counted per sample in a minimum of 30 fields of view. For each microscopic field, two different categories were enumerated: (1) total DAPI-stained cells and (2) cells stained with the specific probe. The counting error, expressed as the percentage of the standard error between replicates, was 2% of the DAPI counts.
The use of different probes targeting Crenarchaeota revealed differences in detection efficiencies (catalyzed reporter deposition-fluorescence in situ hybridization, CARD-FISH, positive) and proportions of Crenarchaeota taking up leucine (MICRO-CARD-FISH positive, see below). Overall, the relative abundance of Crenarchaeota using GI-554 was very similar to that obtained by applying both probes simultaneously (CrenTotal), but, in deep waters, higher than the abundance obtained with the Cren537 probe. De Corte et al. (2009) also report a highly variable detection efficiency for Crenarchaeota using GI-554 and Cren537 in the Eastern Mediterranean Sea. However, these authors found no consistent depth-related trends in the relative abundance of both Cren537-positive and GI-554-positive cells. De Corte et al. (2009) showed that these dissimilarities were due to differences in the coverage of each probe. In this work, not only a higher crenarcheal abundance was obtained using probe GI-554 than the Cren537, but also the proportion of active cells incorporating leucine within this group was always higher (see below).
MICRO-CARD-FISH
MICRO-CARD-FISH (CARD-FISH combined with micro-autoradiography) was performed after the protocol described by Teira et al. (2004). Briefly, samples (10–40 ml) were incubated at in situ temperature with 20 nM final concentration of 3H-Leucine (Leu, Amersham, specific activity=171 Ci mmol−1). Some samples were killed with paraformaldehyde before adding the tritiated leucine and were used as controls. Incubation times varied according to the different depths and ranged between 2 and 24 h. After the incubation, samples were fixed overnight with paraformaldehyde (2% final concentration) at 4 °C, gently filtered onto 0.2 μm polycarbonate filters (Millipore, GTTP, 25 mm diameter), and stored at −80 °C. The filters were then hybridized after the CARD-FISH protocol cited above. The autoradiographic development was conducted by transferring earlier hybridized filter sections onto slides coated with photographic emulsion (type NTB-2, melted at 43 °C for 1 h). Subsequently, the slides were placed in a dark box with a drying agent and exposed at 4 °C for 36–48 h. The slides were developed and fixed using Kodak specifications (Dektol developer [1:1 dilution with Milli-Q water] for 2 min, rinsed with Milli-Q water for 10 s, and fixed for 5 min, followed by a Milli-Q water rinse for 2 min). Cells were counter-stained with the same DAPI mixture used for the CARD-FISH protocol. The silver grains in the autoradiographic emulsion were detected by switching to the transmission mode of the microscope. More than 800 DAPI-stained cells were counted per sample. To enumerate the proportion of Crenarchaeota cells taking up leucine using the two different Crenarchaeota probes, the procedure was repeated using each of the probes (Cren 537 and GI-554) alone or in combination.
DNA sampling, extraction and purification, and fingerprinting of the communities
For DNA fingerprinting of prokaryotic communities, 2–5 l were filtered onto 0.2 μm polycarbonate filters (Millipore, GTTP, 47 mm filter diameter) and the filters stored in microfuge vials in liquid nitrogen for 24 h and then at −80 °C until further processing in the laboratory. DNA extraction was performed using the UltraClean Soil DNA Isolation Kit MoBio kit (MoBIO laboratories, Carlsbad, CA, USA) and the protocol of the manufacturer.
Terminal-restriction fragment length polymorphism for archaeal communities
PCR conditions and chemicals were applied as described in Moeseneder et al. (2001). A total of 1 μl of the DNA extract was used as a template in a 50 μl PCR mixture. For PCR, the Archaea-specific primers 21F-FAM and 958R-JOE were used (Moeseneder et al., 2001). The samples were amplified by an initial denaturation step at 94 °C (for 3 min), followed by 35 cycles of denaturation at 94 °C (1 min), annealing at 55 °C (1 min), and an extension at 72 °C (1 min). Cycling was completed by a final extension at 72 °C for 7 min. The PCR products were run on 1% agarose gel. The gel was stained with a working solution of SYBR Gold and the obtained bands were excised, purified with the Quick gel extraction kit (Genscript, Piscataway, NJ, USA), and quantified using a Nanodrop spectrophotometer. Fluorescently labeled PCR products were digested at 37 °C overnight. Each reaction contained 30 ng of cleaned PCR product, 5 U of tetrameric-restriction enzyme (HhaI), and the respective buffer filled up to a final volume of 50 μl with ultra-pure water (Sigma, St Louis, MO, USA). The restriction enzyme was heat inactivated and precipitated by adding 4.5 μl LPA solution and 100 μl of 100% isopropanol. The samples were kept at room temperature for 15 min followed by centrifugation at 15 000 g for 15 min. Thereafter, the supernatant was discarded and the pellet rinsed with 100 μl 70% isopropanol and precipitated again by centrifugation (15 000 g for 5 min). Subsequently, the supernatant was removed and the sample dried in the cycler at 94 °C for 1 min and stored at −20 °C until further analysis.
The pellet was resuspended in 2 μl of ultra-pure water and the product denatured in 7.8 μl of Hi-Di formamide at 94 °C for 3 min. Each sample contained 0.2 μl GeneTrace 1000 (ROX) marker (Applied Biosystems, Foster City, CA, USA). Fluorescently labeled fragments were separated and detected with an ABI Prism 310 capillary sequencer (Applied Biosystems) run under GeneScan mode (van der Maarel et al., 1998; Moeseneder et al., 1999). The size of the fluorescently labeled fragment was determined by comparison with the internal GeneTrace 1000 (ROX) size standard. Injection was performed electrokinetically at 15 kV and 60 °C for 15 s (adjustable). The output from the ABI Genescan software was transferred to the Fingerprinting II (Bio-Rad, Hercules, CA, USA) software to determine peak area and for standardization using size markers. The obtained matrix was further analyzed with the Primer software (Primer-E, Plymouth Marine Laboratory, Plymouth, UK) to determine similarities of the terminal-restriction fragment length polymorphism fingerprints between samples.
Automated ribosomal intergenic spacer analysis of the bacterial community
Automated ribosomal intergenic spacer analysis was used to analyze bacterial community composition with the primer 1392F and a 5′TET-labeled version of the primer 23S rDNA as described by Fisher and Triplett (1999) and Hewson and Fuhrman (2004). A total of 1 μl of the DNA extract was used as a template in a 50 μl PCR mixture. Thermocycling was preceded by a 3 min heating step at 94 °C, followed by 30 cycles of denaturing at 94 °C (15 s), annealing at 55 °C (30 s), and an extension at 72 °C (3 min). Cycling was completed by a final extension at 72 °C for 9 min. The PCR products were purified with the Quick purification kit (Genscript), and quantified using a Nanodrop spectrophotometer. Purified products were then diluted to 8 ng μl−1 to load a standardized amount for fragment analysis and thereby preventing differences originated from different amounts of loaded DNA. Each sample of the final product was mixed with 10 μl of Hi-Di formamide at 94 °C for 3 min, 0.15 μl CST 300–1800, and 0.15 μl GeneTrace 1000 (ROX) marker (Applied Biosystems). Fragments were discriminated using an ABI Prism 310 capillary sequencer (Applied Biosystems) and the resulting electropherograms were analyzed using the ABI Genescan software. The output from the ABI Genescan software was transferred to the Fingerprinting II (Bio-Rad) software to determine peak area and for standardization using size markers. Peaks contributing <0.09% of the total amplified DNA (as determined by relative fluorescence intensity) were eliminated as considered to be indistinguishable from baseline noise (Hewson and Fuhrman, 2004). The obtained matrix was further analyzed with Primer software (Primer-E) to determine similarities of the automated ribosomal intergenic spacer analysis fingerprints between samples.
Results and discussion
Oceanographic setting
CEs, AEs, and FF stations showed contrasting temperature distributions, with generally lower temperatures in eddies than in FF for both surface and upper mesopelagic waters (except AE1; Figure 2a). The temperature–salinity diagram (Figure 2b) indicates that all the stations shared the same meso- and bathypelagic water mass structure (with the exception of FF2 with a slight influence of Mediterranean Sea Outflow Water). Differences in temperature–salinity properties were only found in the epipelagic layer because of the coastal-ocean-salinity gradient and the mesoscale variability generated by the perturbation of the surface flow by the islands (Figures 2a and b). The CEs CE1 (Figure 3a) and CE2 showed very similar temperatures throughout the water column, whereas the AEs (AE1 and AE2) exhibited temperature differences because of their different stages of development (Figure 2a). AE1 was a typical mature AE, with warm waters mixed down to 200 m (Figure 3b). AE2, close to Gran Canaria Island, was an AE at an early stage of formation, with a warm mixed layer in the upper 60 m, but a strong thermocline underneath.

Vertical profiles of temperature (°C) (a), and temperature–salinity diagram (b) at the six stations (abbreviations as indicated in Figure 1).

Cross section of temperature (°C) of CE1 (a) and AE1 (b) are shown as examples. Black arrows indicate the position of the CTD cast at the core of the eddies.
Differences in prokaryotic structure and function in the epipelagic zone
We observed a marked effect of eddies on the prokaryotic community, generating hotspots of abundance, bulk activity, community composition, and heterotrophic activity at the single-cell level (Figures 4 and 6–8). Generally, the differences between eddy and FF stations were more apparent in the epipelagic layer than in deeper waters.

Distribution of prokaryotic abundance (PA, cells ml−1) and bulk prokaryotic heterotrophic activity (PHA, pmol Leu l−1 h−1) in the epipelagic (‘Epi’, 0–200 m), mesopelagic (‘Meso’, 200–1000 m), and bathypelagic (‘Bathy’, 2000 m) layers grouped in far-fields (FF), anticyclonic eddies (AEs), and cyclonic eddy (CE) stations (a, b), and profiles from surface to 2000 m depth at every sampled station (c, d). Outliers are indicated as open circles. Asterisks indicate variables significantly different (ANOVA test, P<0.05) from FF stations.
The average PA of AE (although not that of CE) was significantly higher than at the FF stations (ANOVA test, P<0.05; Figure 4a). Both AE and CE stations exhibited significantly higher (ANOVA test, P<0.05) bulk PHA than the FF stations (Figure 4c), despite PHA being exceptionally high in the surface layers of FF1 (Figure 4d). In another study carried out in the same region (from the NW African upwelling to the offshore oligotrophic subtropical NE Atlantic), Alonso-Saéz et al. (2007) also found the highest leucine incorporation rates at the offshore stations. They suggested that these high leucine incorporation rates might not reflect proportionally higher PHA, but rather shifts in the leucine-to-carbon conversion factor, which is used to calculate PHA from leucine incorporation rates.
No significant differences (ANOVA test, P>0.05) were detectable, however, in the relative abundance of Bacteria, Crenarchaeota, and Euryarchaeota between eddy and FF stations (Figure 5). The highest relative abundance of Euryarchaeota (14.8% of DAPI stainable cells) was observed in the deep chlorophyll maximum of the well-developed AE1 (Table 1), where also the highest PA was found (Figure 4a). Nevertheless, the relative contribution of Bacteria decreased with depth (Figure 5). In contrast, the relative abundance of Crenarchaeota significantly increased from the epipelagic to mesopelagic layer in all stations.

Distribution of the proportion of (a) Bacteria (Eub (I–III)), (b) Crenarchaeota (hybridized simultaneously with Cren537 and GI-554), (c) Euryarchaeota (Eury806) as percentage of DAPI-stained cells in the epipelagic (‘Epi’, 0–200 m), mesopelagic (‘Meso’, 200–1000 m), and bathypelagic (‘Bathy’, 2000 m) layers grouped in far-fields (FF), anticyclonic eddy (AE), and cyclonic eddy (CE) stations. Outliers are indicated by open circles.
As indicated by the fingerprinting approaches, the bacterial (Figure 6a) and archaeal (Figure 6b) assemblages found within eddies were clearly distinct from the assemblages found in the FF stations only in the epipelagic layer. FF showed very similar bacterial and archaeal structure in the deep chlorophyll maximum. In contrast, although CE1 and CE2 showed a very similar temperature profiles, the bacterial assemblage structures were very different. The contrary occurred at AE1 and AE2 stations that showed contrasting temperature patterns, but a high similarity in bacterial assemblage structure. Therefore, temperature was not the main parameter controlling prokaryotic assemblage structure, but probably other processes (such as grazing, organic and inorganic matter supply, phytoplankton community structure) potentially modified by the presence of the eddies. The total number of operational taxonomic units (OTUs) detected was 104 for bacterial (automated ribosomal intergenic spacer analysis) and 15 for archaeal (terminal-restriction fragment length polymorphism) communities, respectively. The number of archaeal OTUs per sample decreased with depth (from 7±2 to 3±1 in the epipelagic and bathypelagic, respectively), with no significant differences between FF and eddies at any depth layer. In contrast, the number of bacterial OTUs per sample was significantly higher in FF (16) than in eddies (6±1 and 7±3 in the AE and CE stations, respectively) in the epipelagic layer, but not in deeper waters. In deeper waters (meso- and bathypelagic), the number of bacterial OTUs increased, ranging from 16 to 20, 14 to 22, and 16 to 21 for FF, CE, and AE, respectively. About 50% and >30% of the OTUs were shared between both types of eddies and FF for Archaea and Bacteria, respectively. Around 20% of the archaeal and 8% of the bacterial OTUs were present at all the depths sampled, suggesting that the bacterial community was more stratified than the archaeal community.

Non-metric multidimensional scaling (NMDS) plot of the band pattern in (a) bacterial community composition as revealed by automated ribosomal intergenic spacer analysis (ARISA) and (b) archaeal community composition as revealed by terminal-restriction fragment length polymorphism (T-RFLP). Both, band presence alone, and band intensity were used for the statistics, yielding similar results.
The percentages of HNA (Figures 7a and b) and of NADS+ cells (Figures 7c and d) were not significantly different (ANOVA test, P>0.05) between eddies and FF in the three depth layers. The percentage of HNA cells remained fairly constant with depth (Figure 7a), whereas the percentage of NADS+ cells decreased with depth (Figure 7b).

Distribution of the percentage of high nucleic acid (HNA) containing cells and percentage NADS-determined ‘live’ cells in the epipelagic (‘Epi’, 0–200 m), mesopelagic (‘Meso’, 200–1000 m), and bathypelagic (‘Bathy’, 2000 m) layers grouped in far-fields (FF), anticyclonic eddies (AEs), and cyclonic eddies (CEs) stations (a, b), and profiles from surface to 2000 m depth at each station (c, d). Outliers are indicated by open circles. Asterisks indicate variables significantly different (ANOVA test, P<0.05) from FF stations.
The proportion of the prokaryotic community taking up leucine (fraction of leucine-positive cells) (Figure 8a), and the fraction of leucine+ Bacteria (Figure 8b), was higher (ANOVA test, P<0.05) in the epipelagic zone of eddies than at the FF stations (Table 2). Conversely, the percentage of leucine+ Crenarchaota and Euryarchaeota was not significantly different (Figures 8c and Table 2). Although Euryarchaeota were not very abundant, they showed the highest proportion of leucine+ cells (ranging between 48% and 64% of Euryarchaeota) (Table 2).

Distribution of the proportion of (a) the bulk prokaryotic community (DAPI-stained cells), (b) Bacteria (Eub (I–III)), and (c) Crenarchaeota (GI-554) taking up leucine in the epipelagic (‘Epi’, 0–200 m), mesopelagic (‘Meso’, 200–1000 m), and bathypelagic (‘Bathy’, 2000 m) layers grouped in far-field (FF), anticyclonic eddy (AE), and cyclonic eddy (CE) stations. Outliers are shown by open circles. Asterisks indicate variables significantly different (ANOVA test, P<0.05) from FF stations.
The variability found in community structure and activity between eddies and FF probably reflects the accumulation of organic matter at eddy centers and boundary zones in the eddy-field region, as described in earlier studies (Arístegui et al., 2003; Arístegui and Montero, 2005). Frontal structures have been shown to promote the accumulation of organic matter and concomitantly of prokaryotes (Floodgate et al., 1981; Pomeroy et al., 1983; Ducklow, 1988). In particular, Arístegui and Montero (2005) observed that frontal structures between eddy pairs in the Canary region favored the accumulation of bacteria. In addition, Baltar et al. (2009) found dense accumulations of autotrophic and heterotrophic pico- and nanoplankton organisms at eddy-eddy and eddy-filament boundary regions. In addition, the changes found in the prokaryotic activity and community structure may be induced by the higher availability of inorganic nutrients generated by the eddies. The growth of prokaryotic and eukaryotic autotrophs in many aquatic systems is limited by the availability of nitrogen, phosphorus, iron, and silica (for example Elser et al., 1990; Elser et al., 1995). This increased supply of nutrients for surface-water prokaryotes can be produced directly by the upward pumping of deep water (in the CE) and/or the accumulation of planktonic organism (in the center and borders of AE). Owing to the accumulation of microorganisms in AE, the protistan grazing on prokaryotes (an important mechanism of nutrient regeneration in the ocean) may be high. This increased supply of inorganic nutrients may have a stronger effect on prokaryotes than on eukaryotic phytoplankton, because aquatic bacteria are better competitors for phosphorus than eukaryotic algae at low ambient nutrient concentrations (Thingstad et al., 1998). In addition, theory (Klausmeier et al., 2004) and experiments (Sommer, 1994) confirm that different nutrient profiles in the water column select for phytoplankton species with different stoichiometries. This increase in the availability of nutrients may modulate phytoplankton community structure and concomitantly modify prokaryotic assemblage structure and activity (Kelly and Chistoserdov, 2001; Klausmeier et al., 2004). In that sense, Ewart et al. (2008) found a tight relationship between enhanced PHA, phytoplankton biomass, and the specific phytoplankton species, suggesting that phytoplankton community structure was an important factor influencing bacterial activity.
Propagation of effects into deeper layers
The differences in bulk PHA and PA observed in the epipelagic zone between eddy and FF stations propagated, albeit attenuating, into the mesopelagic waters where also higher bulk PHA and PA were detected at the eddy stations (Figure 4). Moreover, some single-cell activity proxies also indicated differences between AE and FF. A significantly higher percentage of NADS+ cells was found in the mesopelagic layer of AE (Figure 7c). However, the proportion of heterotrophically active meso- and bathypelagic prokaryotes was similar in eddies and FF (Figure 8).
In the mesopelagic zone, both CE and AE exhibited significantly higher PA than FF stations (ANOVA test, P<0.05). In the bathypelagic zone, no significant differences in PA were detectable among the different sites. In addition, similar to in the epipelagic zone, eddy stations (AE and CE) exhibited significantly (ANOVA test, P<0.05) higher leucine uptake rates than FF stations in the mesopelagic realm (but not in the bathypelagic) (Figure 4c).
No significant differences (ANOVA test, P>0.05) were detectable in the relative abundance of Bacteria, Crenarchaeota, and Euryarchaeota (Figure 5), the bacterial or archaeal community structures (Figure 6), the percentage of HNA cells (Figures 7a and b), and the proportion of Bacteria, Crenarchaota, or Euryarchaeota taking up leucine (Table 2) between the eddy stations and the FF reference stations in the dark ocean. However, a significantly (ANOVA test, P<0.05) higher percentage of NADS+ cells (Figures 7c and d) was found for the mesopelagic realm of AE as compared with FF (Figures 7c and d), but not in the epi- or bathypelagic layers.
Taken together, these results suggest that only the prokaryotic communities from the epipelagic zone of the eddy stations (and not from deeper waters) are metabolically more active at the single-cell level than those of the FF stations (Figures 6, 7 and 8). The differences found in bulk PA, PHA, and NADS+ cells between eddies and FF in the mesopelagic layer could be related to the increase of the organic matter flux generated by eddies. In a complementary study investigating the flux of particles collected with drifting sediment traps (Alonso-González et al., 2009), the same eddies investigated here were found to enhance particulate organic carbon export with respect to FF stations by a factor of 2–4. These results are in contrast to a study of Maiti et al. (2008) on a mature CE in the lee of Hawaii. They found that, although the eddy was highly productive at the surface, it was not efficient at exporting particulate carbon and nitrogen to deeper waters. In fact, they observed that particle production occurred in the upper 100 m and was rapidly remineralized in the upper 150 m.
Peaks of prokaryotic activity and abundance in the mesopelagic zone
Although PA and metabolism generally decreased with depth, pronounced peaks were sometimes detected in the mesopelagic layer. In particular, PA peaks were observed at 400 m depth at station CE1, coinciding with the deep scattering layer, and in the oxygen minimum zone (ca. 800 m depth) of CE1 and FF2 (Figure 4b). In addition, marked peaks in PHA, HNA, and NADS+ cells were found at several depths of the mesopelagic zone, at different stations (Figures 4b, d, 7b, and d). The two peaks of PA at station CE1 corresponded to large prokaryotes with a high percentage of HNA (compare Figures 4b and 7b), whereas the peak of prokaryotes at the oxygen minimum zone of station FF2 corresponded to cells with the same percentage of HNA as in surface communities (Figure 7b). In the latter peak, a higher proportion of prokaryotes was identified as incorporating leucine (40% leucine+ of DAPI stainable cells) compared with other depths sampled at station FF2 (14–27% leucine+ of DAPI stainable cells). In addition, it coincided with a pronounced peak in NADS+ cells (Figure 7d), and a shift in bacterial (Figure 6a) and archaeal (Figure 6b) community composition. In fact, it is noteworthy that the only deep-water sample clustering with the epipelagic communities of both Bacteria and Archaea was that belonging to the oxygen minimum zone in station FF2 (Figure 6). This might indicate that the prokaryotic community present at this particular depth was more related to that of the deep chlorophyll maximum than to the assemblages of the corresponding depths of other stations.
Mesopelagic hotspots of microbial respiration (Arístegui et al., 2003), prokaryotic nucleic acid content (Baltar et al., 2007), leucine incorporation and leucince/thymidine incorporation ratio (Gasol et al., 2009), dissolved organic carbon (Arístegui et al. 2003), zooplankton biomass, gut fluorescence, and respiration (Hernández-León et al., 2001; Yebra et al., 2005) have been reported in earlier studies for the eddy-field region south of the Canary Islands. Hence, the observed patchiness in mesopelagic activity in these waters seems to be related to the complex hydrographic regime of the region around the Canary Islands.
Relationship between prokaryote viability and single-cell heterotrophic activity throughout the water column
The proportion of viable cells (NADS+ cells) correlated well with the fraction of leucine+ prokaryotic cells (Spearman's R=0.71, P<0.0001, n=26; Figure 9). Furthermore, the percentage of heterotrophically active Bacteria was correlated to the percentage of NADS+ cells (Spearman's R=0.67, P<0.0002, n=26), but no correlation was found for any archaeal group. These results, together with the observed lower relative abundance of both archaeal groups compared with Bacteria, suggest that the fraction of heterotrophically active (leucine+) and viable (NADS+) cells was dominated by Bacteria and not by Archaea throughout the water column. Our results agree with the higher proportion of bacterial cells active in the uptake of leucine, as compared with archaeal cells in the North Atlantic water column (Herndl et al., 2005). As shown in Figure 9, the fraction of viable and heterotrophically active prokaryotes is more similar (that is closer to the 1:1 line) at low than at high values. This indicates that the NADS method likely detects cells that are intact, but not necessarily very active, whereas MICRO-CARD-FISH identifies cells that are active (depending on the concentration of leucine used and on the exposure time), after the prokaryotic ‘physiological structure’ model of del Giorgio and Gasol (2008).

Relationship between the NADS-determined ‘live’ cells and the proportion of active prokaryotic cells taking up leucine (determined by MICRO-CARD-FISH).
Conclusions
Our results show that mesoscale eddies have a differential function in the distribution and function of prokaryotes in the ocean. The largest effects and differences were observed in the upper 1000 m, suggesting that prokaryotic communities are probably linked to the mesoscale heterogeneity, and the increase in productivity and downward flux of organic matter enhanced by eddy action. In general, the effect of AEs was stronger than that of cyclonic ones. Owing to the recognized major function of eddies in ocean circulation, more effort should be put in the future to study the microbial processes within these mesoscale features. This would allow constraining the fate of carbon in the ocean and concomitantly building more accurate models of global biogeochemical cycles.
References
Alonso-González IJ, Arístegui J, Lee C, Calafat A . (2009). Regional and temporal variability of sinking organic matter in the subtropical northeast Atlantic Ocean: a biomarker diagnosis. Biogeosciences, Discussion 6: 11089–11126.
Alonso-Saéz L, Gasol JM, Aristegui J, Vilas JC, Vaqué D, Duarte CM et al. (2007). Large-scale variability in surface bacterial carbon demand and growth efficiency in the subtropical northeast Atlantic Ocean. Limnol Oceanogr 52: 533–546.
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA . (1990). Combination of 16 rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56: 1919–1925.
Arístegui J, Barton ED, Montero MF, Garcia-Muñoz M, Escánez J . (2003). Organic carbon distribution and water column respiration in the NW Africa-Canaries Coastal Transition Zone. Aquat Microb Ecol 33: 289–301.
Arístegui J, Montero MF . (2005). Temporal and spatial changes in plankton respiration and biomass in the Canary Islands region: the effect of mesoscale variability. J Mar Syst 54: 65–82.
Arístegui J, Tett P, Hernández-Guerra A, Basterretxea G, Montero MF, Wild K et al. (1997). The influence of island-generated eddies on chlorophyll distribution: a study of mesoscale variation around Gran Canaria. Deep Sea Res 44: 71–96.
Azam F, Fenchel T, Field JG, Gray JS, Meyer-Reil LA, Thingstad F . (1983). The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 10: 257–263.
Baltar F, Arístegui J, Gasol JM, Hernández-León S, Herndl GJ . (2007). Strong coast—ocean and surface—depth gradients in prokaryotic assemblage structure and activity in a coastal transition zone region. Aquat Microb Ecol 50: 63–74.
Baltar F, Arístegui J, Montero MF, Espino M, Gasol JM, Herndl GJ . (2009). Mesoscale variability modulates seasonal changes in the trophic structure of nano- and picoplankton communities across the NW Africa-Canary Islands transition zone. Prog Oceanogr 83: 180–188.
Benitez-Nelson CR, Bidigare RR, Dickey TD, Landry MR, Leonard CL, Brown SL et al. (2007). Mesoscale eddies drive increased silica export in the Subtropical Pacific Ocean. Science 316: 1017–1021.
Bode A, Barquero S, Varela M, Braun JA, de Armas D . (2001). Pelagic bacteria and phytoplankton in oceanic waters near the Canary Islands in summer. Mar Ecol Prog Ser 209: 1–17.
Cheney RE, Richardson PL . (1976). Observed decay of a cyclonic Gulf Stream ring. Deep Sea Res 23: 143–155.
Daims H, Bruhl A, Amann R, Schleifer KH, Wagner M . (1999). The domain-specific probe EUB338 is insufficient for the detection of all bacteria: developments and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22: 434–444.
De Corte D, Yokokawa T, Varela MM, Agogué H, Herndl GJ . (2009). Spatial distribution of Bacteria and Archaea and amoA gene copy numbers throughout the water column of the Eastern Mediterranean Sea. ISME J 3: 147–158.
del Giorgio PA, Gasol JM . (2008). Physiological structure and single-cell activity in marine bacterioplankton. In: Kirchman DL (ed). Microbial Ecology of the Ocean, 2nd edn, Wiley-Liss: New York. pp 243–298.
Ducklow HW . (1988). Bacterial biomass in warm-core Gulf Stream ring 82-B: mesoscale distributions, temporal changes and production. Deep Sea Res 33: 1789–1812.
Elser JJ, Marzolf ER, Goldman CR . (1990). Phosphorus and nitrogen limitation of phytoplankton growth in the freshwaters of North Ameria: a review and critique of experimental enrichments. Can J Fish Aquat Sci 47: 1468–1477.
Elser JJ, Stabler LB, Hassett RP . (1995). Nutrient limitation of bacterial growth and rates of bacterivory in lakes and oceans: a comparative study. Aquat Microb Ecol 9: 105–110.
Ewart CS, Meyers MK, Wallner ER, McGillicuddy Jr DJ, Carlson CA . (2008). Microbial dynamics in cyclonic and anticyclonic mode-water eddies in the northwestern Sargasso Sea. Deep Sea Res II 55: 1334–1347.
Falcioni T, Papa S, Gasol JM . (2008). Evaluating the flow-fytometric nucleic acid double-staining protocol in realistic rituations of planktonic bacterial death. Appl Environ Microb 74: 1767–1779.
Falkowsky PG, Ziemann DA, Kolber DA, Bienfang PK . (1991). Role of eddy pumping in enhancing primary production in the ocean. Nature 352: 55–58.
Fisher MM, Triplett EW . (1999). Automated approach for ribosomal intergenic spacer analysis of microbial diversity and its application to freshwater bacterial communities. Appl Environ Microbiol 65: 4630–4636.
Floodgate GD, Fogg GE, Jones DA, Lochte K, Turley CM . (1981). Microbiology and zooplankton activity at a front at Liverpool Bay. Nature 240: 133–136.
Gasol JM, Alonso-Sáez L, Vaqué D, Baltar F, Calleja ML, Duarte CM et al. (2009). Mesopelagic prokaryotic bulk and single-cell heterotrophic and community composition in the NW Africa-Canary Islands coastal-transition zone. Prog Oceanogr 83: 189–196.
Gasol JM, Zweifel UL, Peters F, Fuhrman JA, Hagström Å . (1999). Significance of size and nucleic acid content heterogeneity as measured by flow cytometry in natural planktonic bacteria. Appl Environ Microbiol 65: 4475–4483.
González JM, Simó R, Massana R, Covert JS, Casamayor EO, Pedrós-Alió C et al. (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol 66: 4237–4246.
González N, Anadón R, Mouriño B, Fernández E, Sinha B, Escánez J et al. (2001). The metabolic balance of the planktonic community in the North Atlantic subtropical gyre: the role of mesoscale instabilities. Limnol Oceanogr 46: 946–952.
Gregori G, Citterio S, Ghani A, Labra M, Sgorbati S, Brown S et al. (2001). Resolution of viable and membrane-compromised bacteria in freshwater and marine waters based on analytical flow cytometry and nucleic acid double staining. Appl Environ Microbiol 67: 4662–4670.
Harris RP, Boyd P, Harbour DS, Head RN, Pingree RD, Pomroy AJ . (1997). Physical, chemical and biological features of a cyclonic eddy in the region of 61° 10′N 19° 50′W in the North Atlantic. Deep Sea Res I 11: 1815–1839.
Hernández-León S, Gómez M, Pagazaurtundua M, Portillo-Hahnefeld A, Montero I, Almeida C . (2001). Vertical distribution of zooplankton in Canary Island waters: implication for export flux. Deep Sea Res I 48: 1071–1092.
Herndl GJ, Reinthaler T, Teira E, Aken Hv, Veth C, Pernthaler A et al. (2005). Contribution of Archaea to total prokaryotic production in the deep Atlantic Ocean. Appl Environ Microbiol 71: 2303–2309.
Hewson I, Fuhrman JA . (2004). Richness and diversity of bacterioplankton species along an estuarine gradient in Moreton Bay, Australia. Appl Environ Microbiol 70: 3425–3433.
Kelly KM, Chistoserdov AY . (2001). Phylogenetic analysis of the succession of bacterial communities in the Great South Bay (Long Island). FEMS Microbiol Ecol 35: 85–95.
Klausmeier CA, Litchman E, Daufresne T, Levin SA . (2004). Optimal nitrogen-to-phosphorus stoichiometry of phytoplankton. Nature 429: 171–174.
Letelier RM, Karl DM, Abbott MR, Flament P, Freilich M, Lukas R et al. (2000). Role of late winter mesoscale events in the biogeochemical variability of the upper water column of the North Pacific Subtropical Gyre. J Geophys Res Oceans 105: 28723–28739.
Lochte K, Pfannkuche O . (1987). Cyclonic cold-core eddy in the eastern North Atlantic. II. Nutrients, phytoplankton and bacterioplankton. Mar Ecol Prog Ser 39: 153–164.
Maiti K, Benitez-Nelson CR, Rii Y, Bidigare R . (2008). The influence of a mature cyclonic eddy on particle export in the lee of Hawaii. Deep Sea Res II 55: 1445–1460.
Massana R, Murray AE, Preston CM, DeLong EF . (1997). Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl Environ Microbiol 63: 50–56.
Mathis JT, Pickart RS, Hansell DA, Kadko D, Bates NR . (2007). Eddy transport of organic carbon and nutrients from the Chukchi Shelf: impact on the upper halocline of the western Arctic Ocean. J Geophys Res 112: C05011.
McGillicuddy Jr DJ, Robinson AR, Siegel DA, Jannasch HW, Johnson R, Dickey TD, et al. (1998). Influence of mesoscale eddies on new production in the Sargasso Sea. Nature 394: 263–266.
Moeseneder MM, Arrieta JM, Muyzer G, Winter C, Herndl GJ . (1999). Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl Environ Microbiol 65: 3518–3525.
Moeseneder MM, Winter C, Arrieta JM, Herndl GJ . (2001). Terminal restriction fragment length polymorphism (T-RFLP) screening of a marine archaeal library to determine the different phylotypes. J Microbiol Meth 44: 159–172.
Morán XAG, Taupier-Letage I, Vázquez-Domínguez E, Ruiz S, Arin L, Raimbault P et al. (2001). Physical-biological coupling in the Algerian Basin (SW Mediterranean): influence of mesoscale instabilities on the biomass and production of phytoplankton and bacterioplankton. Deep Sea Res I 48: 405–437.
Mouriño-Carballido Jr B, McGillicuddy DJ . (2006). Mesoscale variability in the metabolic balance of the Sargasso Sea. Limnol Oceanogr 51: 2675–2689.
Pomeroy LR, Atkinson LP, Blanton JO, Campbell WB, Jacobsen TR, Kerrick KH et al. (1983). Microbial distribution and abundance in response to physical and biological processes on the continental shelf of southeastern USA. Cont Shelf Res 2: 1–20.
Smith DC, Azam F . (1992). A simple, economical method for measuring bacterial protein synthesis rates in seawater using 3H-leucine. Mar Microb Food Webs 6: 107–114.
Sommer U . (1994). The impact of light intensity and day length on silicate and nitrate competition among marine phytoplankton. Limnol Oceanogr 39: 1680–1688.
Tarran GA, Zubkov MV, Sleigh MA, Burkill PH, Yallop M . (2001). Microbial community structure and standing stocks in the NE Atlantic in June and July of 1996. Deep Sea Res II 48: 963–985.
Teira E, Reinthaler T, Pernthaler A, Pernthaler J, Herndl GJ . (2004). Combining catalyzed reporter deposition-fluorescence in situ hybridization and microautoradiography to detect substrate utilization by Bacteria and Archaea in the deep ocean. Appl Environ Microbiol 70: 44411–44414.
Thingstad TF, Zweifel UL, Rassoulzadegan F . (1998). P limitation of heterotrophic bacteria and phytoplankton in the northwest Mediterranean. Limnol Oceanogr 43: 88–94.
Thyssen M, Lefevre D, Caniaux G, Ras J, Fernandez CI, Denis M . (2005). Spatial distribution of heterotrophic bacteria in the northeast Atlantic (POMME study area) during spring 2001. J Geophys Res 110: C07S16, doi:10.1029/2004JC002670.
van der Maarel MJEC, Artz RRE, Haanstra R, Forney LJ . (1998). Association of marine Archaea with the digestive tracts of two marine fish species. Appl Environ Microbiol 64: 2894–2898.
van Haren H, Millot C, Taupier-Letage I . (2006). Fast deep sinking in Mediterranean eddies. Geophys Res Lett 33: L04606:10.1029/2005GL025367.
Yebra L, Almeida C, Hernández-León S . (2005). Vertical distribution of zooplankton and active flux across an anticyclonic eddy in the Canary Island waters. Deep Sea Res I 52: 69–83.
Zhang Y, Sintes E, Chen J, Zhang Y, Dai M, Jiao N et al. (2009). Role of mesoscale cyclonic eddies in the distribution and activity of Archaea and Bacteria in the South China Sea. Aquat Microb Ecol 56: 65–79.
Acknowledgements
This research was supported by two grants of the Spanish Ministry of Education and Science to JA (Oceanic Eddies and Atmospheric Deposition—RODA, CTM 2004-06842-C03/MAR, and Shelf–Ocean Exchanges in the Canaries–Iberian Large Marine Ecosystem-CAIBEX, CTM 2007-66498-C02), a grant of the Earth and Life Science Division of the Dutch Science Foundation (ALW-NWO; ARCHIMEDES project, 835.20.023) to GJH, and a predoctoral Fellowship of the Spanish Ministry of Education and Science (AP2005-3932) to FB. IL and JMG were also supported by project MODIVUS (CTM2005-04795/MAR). The work was carried out within the frame of the EU ‘Networks of Excellence’ MarBef and EurOceans. We acknowledge the insightful comments of two anonymous reviewers, which helped to improve the manuscript. We thank the captain and crew of R/V Hespérides for their help during work at sea. We also thank A Marrero-Díaz for providing the temperature cross-sections, and AM Caballero-Alfonso for her help during sampling and DNA extraction.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baltar, F., Arístegui, J., Gasol, J. et al. Mesoscale eddies: hotspots of prokaryotic activity and differential community structure in the ocean. ISME J 4, 975–988 (2010). https://doi.org/10.1038/ismej.2010.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.33
Keywords
This article is cited by
-
Dynamics in Bacterial Community Affected by Mesoscale Eddies in the Northern Slope of the South China Sea
Microbial Ecology (2022)
-
Depth-Dependent Variables Shape Community Structure and Functionality in the Prince Edward Islands
Microbial Ecology (2021)
-
Bacterial community variations in the South China Sea driven by different chemical conditions
Ecotoxicology (2021)
-
Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities
Communications Biology (2021)
-
Reducing the arbitrary: fuzzy detection of microbial ecotones and ecosystems – focus on the pelagic environment
Environmental Microbiome (2020)
