
Abstract
The Brazos-Trinity Basin on the slope of the Gulf of Mexico passive margin was drilled during Integrated Ocean Drilling Progam Expedition 308. The buried anaerobic sediments of this basin are largely organic-poor and have few microbial inhabitants compared with the organic-rich sediments with high cell counts from the Peru Margin that were drilled during Ocean Drilling Program Leg 201. Nucleic acids were extracted from Brazos-Trinity Basin sediments and were subjected to whole-genome amplification and pyrosequencing. A comparison of the Brazos-Trinity Basin metagenome, consisting of 105 Mbp, and the existing Peru Margin metagenome revealed trends linking gene content, phylogenetic content, geological location and geochemical regime. The major microbial groups (Proteobacteria, Firmicutes, Euryarchaeota and Chloroflexi) occur consistently throughout all samples, yet their shifting abundances allow for discrimination between samples. The cluster of orthologous groups category abundances for some classes of genes are correlated with geochemical factors, such as the level of ammonia. Here we describe the sediment metagenome from the oligotrophic Brazos-Trinity Basin (Site 1320) and show similarities and differences with the dataset from the Pacific Peru Margin (Site 1229) and other pyrosequenced datasets. The microbial community found at Integrated Ocean Drilling Program Site 1320 likely represents the subsurface microbial inhabitants of turbiditic slopes that lack substantial upwelling.
Similar content being viewed by others


Introduction
The deep marine biosphere is thought to host a large component of the world's microbial population, yet little is known about these subsurface cells (Whitman et al., 1998). The microbial inhabitants of the sediment have been explored using many techniques, including but not limited to clone library analysis, lipid extractions, microscopy and quantitative QPCR (Schippers et al., 2005; Biddle et al., 2006; Inagaki et al., 2006; Lipp et al., 2008). The majority of these studies have been conducted on sediments under significant upwelling zones along active accretionary margins, in places where total organic carbon( content is high (1–5%) and geochemical profiles of methane and sulfate indicate active anaerobic microbial communities (D’Hondt et al., 2004). However, many marine sediments are poor in organic carbon (0.03–1% of the total organic carbon). With the proposed hypothesis that many of the subsurface microbes, particularly the Archaea, are dependent on organic carbon for their metabolism (Biddle et al., 2006; Lipp et al., 2008), it stands to reason that sediments with differing organic carbon contents have distinct microbial communities (Inagaki et al., 2006; Teske, 2006; Fry et al., 2008; Lipp et al., 2008). Ongoing studies of open-ocean sediment from the Equatorial Pacific (D’Hondt et al., 2004), Peru Basin (Sørensen et al., 2004) and the South Pacific gyre (D’Hondt et al., 2009; Durbin and Teske, 2010) aim to help elucidate the microbial communities present in organic-poor, open-ocean subsurface settings. However, only a single PCR-based study of the microbial communities found in organic-poor turbiditic margin sediments, such as Integrated Ocean Drilling Program (IODP) Site 1320, has been conducted (Nunoura et al., 2009). The study of this organic-poor anaerobic margin sediment using a quantitative technique that provides phylogenetic information, such as pyrosequencing a metagenome, could provide a counterpoint to the organic-rich sediments previously studied and thus provide a more complete view of subsurface marine microbiology than has been previously observed.
One of the most studied regions of marine sediment is that of the Peru Margin. Drilled in 2002 during Ocean Drilling Program (ODP) Leg 201, these sediments were found to have active geochemical cycling driven by microbes and microbial populations ranging from 106 to 109 cells per cm–3 sediment (D’Hondt et al., 2004). Cores from one site in particular, Peru Margin Site 1229, have been subject to numerous studies, including a metagenomic analysis (Biddle et al., 2008). During the initial screening of this metagenome, major differences were seen between this sediment environment and most terrestrial and pelagic metagenomes (Biddle et al., 2008), but few differences, based on gene content and phylogenetic associations, were seen between sediment depth horizons (Teske and Biddle, 2008). Owing to the high microbial activity of these sediments, the Peru Margin metagenome may not be representative of all subsurface marine sediment microbes and may only represent the organic-rich end of the spectrum of sediments.
Sediments from the Brazos-Trinity Basin, part of the Gulf of Mexico passive margin, were drilled during IODP Leg 308. Sediments from this leg were oligotrophic (low organic carbon content), because of a low sedimentation rate resulting in an average total organic carbon of 0.53 wt% (Expedition 308 Scientists, 2005). Site 1320, in the lower part of the basin, has microbial populations ranging from 104 to 106 cells per cm–3 sediment, resulting in orders of magnitude fewer cells than Site 1229 (Nunoura et al., 2009). However, this site has geochemical gradients similar to Site 1229, indicating that sulfate reduction, methane oxidation and methanogenesis should occur at specific depths throughout the sediment column (Nunoura et al., 2009). Additionally, the microbial groups detected by 16S rRNA gene clone library analysis are quite similar to those found in other deep subsurface sediments. The bacterial clone libraries are dominated by the Chloroflexi phylum, followed by proteobacterial lineages. The archaeal clone libraries consist mostly of members of the Miscellaneous Crenarchaeotal Group, followed by common subsurface lineages known as the Deep Sea Archaeal Group (also known as Marine Benthic Group B) and South-African Gold Mine Euryarchaetoal Group (Nunoura et al., 2009). These phylum-level groups are quite similar to those found at Site 1229 (Parkes et al., 2005; Biddle et al., 2006, 2008). A few noticeable differences are seen between sites, with Spirochetes, Deltaproteobacteria and Japan Sea 1 bacterial groups detected in larger percentages in shallow depths at Site 1320 than at Site 1229 (Parkes et al., 2005; Nunoura et al., 2009).
The clone library differences between the Peru Margin and Brazos-Trinity Basin microbial communities justified a metagenomic analysis. A metagenomic analysis allows for all genes in an environment to be sequenced, whereas clone library studies are restricted to mostly ribosomal genes and few known functional genes. A single metagenomic analysis provides both taxonomic and functional information, providing the widest view of microbial community structure and genome repertoire in the environment (Hugenholtz and Tyson, 2008) and may provide a relatively unbiased view of diversity (Biddle et al., 2008). Additionally, a metagenome from this site allows for comparative studies between other environments sampled in the same way.
The low cell abundance in the Brazos-Trinity sediments, nearly two orders of magnitude lower than the Peru Margin, meant that only shallow sediment depths were targeted for metagenomic study as the previous 16S rRNA clone library study was most successful in amplifying DNA from depths shallower than 22 meters below seafloor (mbsf). DNA was extracted from 8 mbsf in the Brazos-Trinity basin and amplified using multiple displacement whole genome amplification under monitored amplification conditions and sequenced for the total genome content. Here we present the analysis of this new metagenome, using the existing Peru Margin metagenome and other pyrosequenced metagenomes as comparative samples.
Materials and methods
Site description
IODP Expedition 308 Site 1320 (27°18.0809′N 94°23.2537′W) is in the Brazos-Trinity Basin IV in the Gulf of Mexico in 1470 m water depth. A total of 299 m of sediment were retrieved for IODP Expedition 308 Hole U1320A. Sediment was sectioned on-ship and immediately placed into low-permeability bags and flushed with N2 gas (Expedition 308 Scientists, 2005). Core 1320A had some of the highest cell counts (Expedition 308 Scientists, 2005), and, at approximately 10 mbsf, the highest number of prokaryotic rRNA genes within this expeditions sample set (Nunoura et al., 2009). Frozen (−80 °C) repository samples from IODP Expedition 308 Hole U1320A section 2H3 (8 mbsf) and 4H5 (30 mbsf) were used in this study. Geochemical data on the samples were compiled from ship-board data (Supplementary Table S1; Expedition 308 Scientists, 2005).
Nucleic acid extraction
A phenol extraction was performed using the following protocol (adapted from Biddle et al., 2006). In a 50-ml conical tube, 5 ml Tris-buffered phenol (pH 8), 5 ml 5 × extraction buffer (250 mM sodium acetate, 50 mM EDTA, pH adjusted to 8), 0.5 ml 20% SDS, 2 g 0.1-μm sterile glass beads and 5 g sediment were combined and vortexed for 1 min. The samples were then centrifuged at 5000 × g for 5 min and the aqueous phase was removed into a new tube. In all, 5 ml of phenol:chloroform:isoamyl alcohol, pH 8, was added and the mixture was briefly vortexed. The sample was then recentrifuged as above and the aqueous phase was again removed to a new tube. Then 5 ml of chloroform was added, the sample was vortexed briefly, centrifuged and the aqueous phase removed. To precipitate DNA, 2.5 ml 7.5 M ammonium acetate and 5 ml isopropanol were added. After mixing the sample by gentle inversion, it was incubated at room temperature for 30 min, then centrifuged at 5000 × g for 30 min. All liquid was removed and the invisible pellet was washed with 70% ethanol and air dried. DNA was resuspended in 20 μl PCR grade water. Tests showed this extract contained polymerase inhibitors and required a cleaning step of gel purification. For gel purification, 10 μl of sample volume was mixed with 2 μl loading buffer, and was run on a 1% low melting point, surface tension TAE gel at 100 V for 10 min. When the gel was stained with SYBR gold (Invitrogen, Carlsbad, CA, USA), no bands were visible; however, the space between the sample wells and the dye front was excised and subjected to gel purification using the digestive enzyme Gelase (Invitrogen) as per the manufacturer's instructions. As a contamination control, a gel slice was excised from a lane that had been loaded with only loading dye. The precipitated DNA was resuspended in 2.5 μl PCR grade water.
Whole genome amplification (WGA) and contamination control
After the gel purification, the entire sample was subjected to phi29 whole genome amplification using the RepliG Midi kit (Qiagen, Valencia, CA, USA). Manufacturer's instructions were followed, with a few exceptions: the water added to the reaction included SYBR green I (Invitrogen) at 50 × concentration and the incubation was visualized by an MX3500P QPCR machine (Stratagene, La Jolla, CA, USA) every 10 min. The total reaction time was only 90 min, after which the reaction was stopped by heating to 65 °C for 3 min (Supplementary Figure S1). DNAs were diluted 1:10 and 1 μl was used for amplification of bacterial and archaeal 16S rRNA genes. Amplification of these genes was seen for the 2H3 sample, but neither for the negative controls (gel purification control and water control) nor for the 4H5 sample. DNA present in the 4H5 sample may have been unable to amplify within the short amplification time; however, attempts to amplify for longer times resulted in amplification within the negative controls. As such, only the 2H3 sample amplified for this short time was used in further experiments.
Pyrosequencing
Amplified DNA from Site 1320A 2H3, approximately 100 ng, was sent to the Pennsylvania State University Center for Genomic Analysis, where pyrosequencing was performed according to company protocol on a 454 Life Sciences GSFLX system (Branford, CT, USA). Sequences from this project were deposited in the NCBI Short Read Archive (SRA009400).
Sequence analysis
The resulting sequence from Site 1320 (Brazos-Trinity Basin, 8 mbsf), the existing sequence from the Peru Margin (NCBI accession code SRA001015; Peru Margin depths 1, 1*, 16, 32 and 50 mbsf with 1* denoting a parallel sample from 1 mbsf without WGA) were processed as follows. To detect homology, sequences from all environments were searched against the NCBI nonredundant protein database using BLASTX. Hits to the NR database required an e-value <1e−5. Parameters for all BLASTX searches were ‘−b 50–e 0.00001−W 4.’ Sequences with matches to the NR database were assigned to the taxonomic clade of their corresponding best hit. The taxonomic identity of each database entry was determined using the NCBI taxonomy (public hierarchy files names.dmp, nodes.dmp) with corresponding accession or gi numbers. Sequences with matches to eukaryotes were removed before additional analyses.
Additional BLASTX analyses were performed with these data and comparative datasets, Red/Black Soudan Mine and Lean/Obese Gut Flora (all comparative datasets were retrieved from the MG-RAST server, http://metagenomics.nmpdr.org/; Metagenome IDs: 4440281.3, 4440282.3, 4440463.3, 4440464.3), against the NCBI clusters of orthologous groups (COG) database (Tatusov et al., 2003) (obtained in 2009, http://www.ncbi.nlm.nih.gov/COG/) to create functional profiles for each sample (e-value <1e−5). Sequences with matches to the COG database were assigned to general functional groups based on the best BLASTX hit. Some COGs are associated with multiple general functions (for example, COG0304 [IQ] is associated with lipid metabolism [I] and secondary metabolite biosynthesis [Q]).
Taxonomic composition estimation
Sequences with COG hits were searched for 31 specific COGs associated with high-quality phylogenetic markers. In contrast to phylogenetic profiles created using rRNA gene hits (for example, the 16S rRNA gene), these markers are single-copy protein-coding genes, and thus permit a less-biased quantitative description of the taxonomic composition of a microbial community (Ciccarelli et al., 2006; von Mering et al., 2007). Hits to these markers were collected for each sample to form taxonomic profiles.
Statistical analysis
Samples were clustered according to taxonomic composition profiles and COG profiles using a distance function of D=(1−∣Pearson's correlation coefficient∣) and complete-linkage clustering. Before clustering, functional COG profile values within each category were normalized across samples by computing the difference from the mean and dividing by the corresponding standard deviation. The associated dendrograms were visualized using the heatmap_2 function in the R library package ‘Heatplus.’ To detect differentially abundant metagenomic features between each Peru Margin Site 1229 depth horizon and the Brazos-Trinity Basin sample, the Metastats (White et al., 2009) statistical methodology was modified to include comparisons between two single samples. All sample comparisons of functional abundances employed the χ2 test. Owing to the sparse observations associated with some of the taxonomic abundances, sample comparisons of taxonomic composition employed Fisher's exact test. Correlations between the metadata and feature abundances were further examined using Pearson's product-moment correlation coefficient. Features included the relative abundance of each phylum in the taxonomic profile, and the relative abundance of each COG functional category. Metadata variables included sampling depth, site location, and concentrations of ammonium (NH4), sulfate (SO4) and iron (Fe) (Tables 1 and Supplementary Table S1) (Expedition 308 Scientists, 2005).
Results
Nucleic acids were extracted from the 1320A 2H3 (8 mbsf) Brazos-Trinity Basin sample, but not from the considerably deeper sample from 4H5 (30 mbsf). The amplification of this extracted DNA was detected by observing the whole genome amplification reaction on a QPCR machine where a clear amplification signal was seen from the 2H3 sample, but not the negative control (Supplementary Figure S1). No amplification was seen by WGA or PCR from the 4H5 sample. Pyrosequencing was performed on the 2H3, 8 mbsf, sample and a total of 546 127 reads were retrieved with an average sequence length of 193 basepairs (bp), for a total of just over 105 Mbp. Sequences from this Brazos-Trinity Basin sample, along with the existing Peru Margin samples, were first analyzed by BLASTX and hits were binned according to their associated taxonomy (Table 1). To aid in the analysis of the sediment microbial population, all genes identified as eukaryotic were removed from further analysis. The Brazos-Trinity Basin sample contained 1.9% eukaryotic-associated hits (Table 1). Half of these matches were to human sequences, fewer matched mouse, pufferfish or other eukaryotes (data not shown). Although these genes may actually be homologs from prokaryotes and may be misannotated, the inability to prove this possibility required they be removed from further study. The metagenomes contain few marker genes for specific microbial metabolisms, a phenomenon also found in the initial analysis of Peru Margin metagenome data (Biddle et al., 2008). As specific genes were insufficiently abundant for statistical analysis, the genetic comparisons of samples were based on COG association and taxonomic association, that is, bacterial and archaeal phylum-level gene identification.
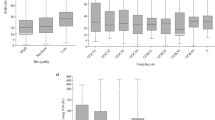
The taxonomic associations according to BLAST are similar for all samples (Figure 1a), in that the major phylogenetic groups of Bacteria and Archaea are consistently represented. In the Brazos-Trinity 8 msbf sample, the Proteobacteria have the largest assigned number of genes (24.9%), followed by the Firmicutes (23%), Euryarchaeota (19.8%) and Chloroflexi (10%). These groups account for 77.7% of all assigned reads. In contrast, as described previously, the 50 mbsf sample from Peru Margin has the maximum number of reads assigned to Euryarchaetoa (24.5%), followed by Proteobacteria (18%), Firmicutes (18%) and Chloroflexi (15.7%), resulting in a total of 76.2% for these four dominating groups. A large percentage of BLAST-identified reads were assigned to COG categories (72–85% among samples). When single-copy genes often used as phylogenetic markers were sampled, the taxonomic profiles for each sample shifted (Figure 1b) (von Mering et al., 2007). Most notably, in the Brazos-Trinity sample there is a large increase in the representation of Spirochetes. Also, the representation of Crenarchaeota increased in all samples as the representation of Proteobacteria decreased. Using the Fisher's exact test, the single-copy COG phylogenetic markers from the Brazos-Trinity 8 mbsf sample are significantly enriched in Spirochetes and depleted in Chloroflexi compared with the Peru Margin (P<1e−06 for all comparisons). These results are consistent with the previous clone library study, which shows that Spirochetes were found in greater abundance in the Brazos-Trinity Basin than had been found at the Peru Margin (Parkes et al., 2005; Nunoura et al., 2009).

Phylogenetic associations of all BLAST hits (nr database) (a) and only single copy protein-encoding genes (b) for the Brazos-Trinity Basin and Peru Margin datasets. (a) The BT basin, PM 1*, PM1, PM16, PM32 and PM50 as percentages of the total reads (respectively: 121 681, 6726, 7031, 3052, 3919, 5485) annotated. (b) The same samples with only reads annotated as single-copy protein-encoding genes (respectively: 1836, 164, 163, 77, 96, 184).
To quantify the diversity of each sample, the taxonomic composition profiles were used to compute the Chao1 estimator and the Shannon diversity index (Table 1). We used genus-level assignments to compute Shannon and Chao estimates as a lower limit of measured diversity, as species-level information is not available. Across all samples, the Shannon indices were remarkably consistent, whereas the computed Chao1 estimate for the Brazos-Trinity 8 mbsf sample was more than twice that of the Peru Margin samples. However, this result is likely due to the sampling depth of the BT sample compared with the Peru Margin samples, as Chao1 estimates always tend to be higher with increased sampling frequency. Thus, the diversity of these environments detected through metagenomics is notably consistent despite differences in relative phylogenetic abundances.
χ2 tests were run to detect significantly different COG categories between the Brazos-Trinity Basin and the Peru Margin. COG categories that host genes for higher structural complexity were not significantly different, such as nuclear structure [Y] and extracellular structures [W] (Supplementary Table S2). Other categories were significantly different between the two data sets, such as carbohydrate metabolism [G], lipid transport [I], transcription [K], translation [J] and posttranslation categories [O] (P<1e−4 for all corresponding comparisons). To examine overall similarity among the samples, COG abundance profiles were subjected to hierarchical clustering analysis based on Pearson's correlation coefficients (Figure 2). Using this representation, we see from the clustering that the COG distribution in the Brazos-Trinity Basin has a distant relationship with the COG distributions from the Peru Margin, as COGs are not equally distributed throughout these subsurface samples. The Brazos-Trinity Basin and Peru Margin microbial communities are distinct from each other, likely because of geographic separation. Further, the depth horizons of the Peru Margin site can be classified into three different geochemical regimes: at 50 mbsf, where methane is generated; at 32 and 16 mbsf, where sulfate is reduced; at 1 mbsf, where also sulfate is reduced, but it most likely contains less recalcitrant organic matter (Meister et al., 2005). This association was not apparent in earlier analyses of these data (Biddle et al., 2008).

Hierarchical clustering based on COG category abundances. Values within each category are normalized across samples (see Materials and methods). COG category codes are as follows: [U], intracellular trafficking and secretion; [G], carbohydrate transport and metabolism; [I], lipid transport and metabolism; [R], general function prediction only; [D], cell cycle control, mitosis and meiosis; [H], coenzyme transport and metabolism; [B], chromatin structure and dynamics; [P], inorganic ion transport and metabolism; [O], posttranslational modification, protein turnover, chaperones; [J], translation; [A], RNA processing and modification; [L], replication, recombination and repair; [C], energy production and conversion; [M], cell wall/membrane biogenesis; [Q], secondary metabolites biosynthesis, transport and catabolism; [V], defense mechanisms; [E], amino acid transport and metabolism; [K], transcription; [N], cell motility; [T], signal transduction mechanisms; [F], nucleotide transport and metabolism; and [S], function unknown. The asterisk denotes the unamplified 1 mbsf Peru Margin sample.
In contrast to clustering by COG category abundance, hierarchical clustering based on taxonomic abundances indicates that the deeper Peru Margin samples at 16, 32 and 50 mbsf cluster separately from the two shallow samples, the 1 mbsf Peru Margin and the 8 mbsf Brazos-Trinity samples (Figure 3). Variability in the levels of dominant prokaryotes, including Euryarchaeota, Proteobacteria, Firmicutes and Chloroflexi, appears to drive this clustering. The dominance of these few groups can clearly be seen over all other taxa, showing their potential importance to the subsurface community (Figure 3).

Hierarchical clustering based on taxonomic associations of BLAST hits to single-copy phylogenetically informative COGs. The colored scale indicates the proportion of matches to each category. The asterisk denotes the unamplified 1 mbsf Peru Margin sample.
To test whether there are genetic associations with measured geochemistries, as suggested by the clustering analysis, R2 Pearson correlation coefficients were determined between COG categories and geochemical measurements (Supplementary Tables S1 and S2). This analysis shows that the abundance of sequence reads assigned to the energy production COG is highly correlated with several metadata features including depth, ammonia and sulfate (r2 values of 0.77, 0.90 and 0.84, respectively) (Supplementary Table S3). There are strong correlations detected between levels of ammonia and the relative abundance of several COG categories, including a positive correlation of energy conversion/production and post-translational modification, and a negative correlation of membrane biogenesis and cell motility with increasing ammonia levels (Figure 4). Other metadata features correlated well, such as the presence of genes classified within the category of inorganic ion transport and the level of manganese (Supplementary Table S3); however, it is currently difficult to separate such trends from the classic diagenetic profile of a sediment column.

Correlation of COG data with metadata: genes related to cell motility (a), membrane biogenesis (b), post-translational modification (c) and energy production/conversion (d) are well correlated with levels of ammonia (r2 values of 0.72, 0.65, 0.72 and 0.90, respectively). Data points in each graph, from left to right, correspond to samples BT basin (filled square, 998 μM ammonia), Peru Margin 1 mbsf amplified (diamond, 2419 μM ammonia), Peru Margin 16 mbsf (triangle, 2805 μM ammonia), Peru Margin 32 mbsf (circle, 4078 μM ammonia) and Peru Margin 50 mbsf (open square, 5000 μM ammonia).
Using a principal component analysis of COG category abundance, the sediment metagenomes were compared with other pyrosequenced metagenomes from public databases (Figure 5). In this plot, each principal component is a linear combination of COG category relative abundances. The previous clustering used the normalized measure of COG category abundance across samples to group the samples (Figure 2); by this measure, the Brazos-Trinity sample was clustered as distinct from the Peru Margin samples. In this new analysis, which relies on the relative abundance of COG categories, the COGs from the Brazos-Trinity sample are also distinct from those from the Peru Margin (Figure 5). The shallow Peru Margin samples bridge the gap between the highly distinct deep Peru Margin samples and the Brazos-Trinity Basin sample. Further, the sediment samples are compared with samples from the Soudan Mine, a terrestrial deep biosphere sample (Edwards et al., 2006), and samples from lean and obese human gut flora (Turnbaugh et al., 2008). The Brazos-Trinity samples are most similar to the Soudan Mine samples. Completely distinct from all deep environmental samples are the samples from lean and obese human gut flora (Figure 5). Taxonomic comparisons of phylogenetically informative COGs for these samples show that taxon differences alone do not drive the comparisons of COG categories, as the mine and gut samples look more taxonomically similar to each other compared with the sediment samples (Supplementary Figure S2).

Principal component analysis of COG category relative abundance of the Brazos-Trinity Basin (circle); 50, 30 and 16 mbsf Peru Margin samples (dark triangles); 1 mbsf Peru Margin samples (gray triangles); Soudan Mine samples (diamonds); and lean and obese human gut samples (gray circles). Percentages on axes denote the amount of variance explained by each principal component.
Discussion
The microbial community observed buried 8 mbsf at Brazos-Trinity Basin on the slope of the Gulf of Mexico passive margin lives in organic-poor anaerobic sediments. The degree to which the studied community appears similar to those observed in the subsurface of the organic-rich Peru Margin probably gives the best insight into which microbial groups are ubiquitous in anaerobic subsurface marine sediments. However, where the subsurface microbiology of the Brazos-Trinity Basin differs from the Peru Margin provides new insight into a subsurface microbial community composition that might be generally found in turbiditic slopes when substantial upwelling is not present. By comparing the metagenomes of sediments that are similar because of the lack of oxygen, yet different in their levels of total organic carbon, we are able to observe relationships between microbes and their environments. These data must be interpreted with caveats, because of the amplification of sample and lack of replicates for the Brazos-Trinity sample. However, taken as a whole, with the existing work (Nunoura et al., 2009), interesting insights can be made into these samples.
Upon the initial evaluation of the Peru Margin metagenome, it appeared to have relatively consistent gene content at different depths, despite drastic changes in geochemical conditions (Biddle et al., 2008). Now, using the Brazos-Trinity Basin Site 1320 anaerobic sediment as a comparative sample and applying statistical analyses, trends are being seen with gene content categories and taxonomic associations. Similarities in the taxonomic spectrum of major microbial groups link these geographically distant sediments, potentially because of the anaerobic nature of the sediments, despite the changes in total organic carbon content. However, differences are seen that are potentially due to biogeography and biogeochemistry. Additionally, the association of COG categories seen with increasing levels of ammonia in the sediments is intriguing (Figure 4); however, its meaning is currently unclear. Many of the COG categories have similar trends, with the chemical species usually seen changing in a diagenetic profile (Supplementary Table S3), such as a decrease in genes for energy production and conversion correlating with decreasing levels of sulfate (r2=0.837, Supplementary Table S3). The correlations with depth alone are slightly lower; the best depth correlation appears in genes linked to energy production and conversion (r2=0.769) (Supplementary Table S3). The correlations of COG category abundance to depth, age or chemistry of sediment are suggestive, but further studies and a wider range of sampling sites are required to constrain the linkages of geochemistry, microbiology, age and depth of sediments, and to establish or disprove their mutual interdependence.
The Brazos-Trinity dataset shows that the basic representation of a deep sediment metagenome is composed of four main microbial groups, the Euryarchaeota, Proteobacteria, Chloroflexi and Firmicutes. These main groups are seen by the analysis of all genes throughout all samples, including one not subjected to WGA (Figure 1a). The hierarchical clustering analysis suggests that these phylogenetically defined groups influence the similarities and differences of sediment metagenomes (Figure 3). Yet, these clustering patterns are different for COG category abundances across the same samples (Figure 2). This discrepancy between phylogenetic and COG category-based clustering patterns is intriguing and could be interpreted as circumstantial evidence for systematically diverging genome content and COG category abundance within the mostly uncultivated, dominant microbial groups of deep marine sediments. Additionally, while the Euryarchaeota are identified as a major group by the metagenome, 16S rRNA gene studies have indicated that Crenarchaeota are the dominant Archaea (Nunoura et al., 2009). The discrepancy between total archaeal gene identification and small subunit ribosomal identification in sediments has been documented previously (Biddle et al., 2008), and the databases may still not contain sufficiently close templates for a robust taxonomic identification of the overall crenarchaeotal gene content in the sediment. Also, the singular instance of increased Spirochetes in the Brazos-Trinity Basin sample seen by ribosomal protein identification (Figure 1b), while in support of the clone library findings from this site (Nunoura et al., 2009), may be exaggerated in the metagenome as certain genes may be randomly overamplified during whole genome amplification, despite controls to avoid this; unfortunately, multiple samples are not available for comparison. These caveats serve as a reminder that genome amplification techniques that are intended to improve microbial detection should be used with caution (Lipp et al., 2008) and that the taxonomic associations of a metagenome cannot be the only evidence for the microbes in an environment (Biddle et al., 2008). In this study, the continual analysis of both the amplified and unamplified 1 mbsf Peru Margin sample allows for a measure of WGA-induced noise to be evaluated for each individual test. The clustering analyses run in this study always produce these two samples as being most closely related compared with the other sediment samples processed in the same way, suggesting that introduced noise is a smaller factor than true sample-to-sample differences.
An interesting phenomenon seen in this new analysis is that the 1 mbsf sample from the Peru Margin and the 8 mbsf sample from the Brazos-Trinity Basin appear distinct from the deeper samples according to taxonomic associations (Figure 3). This reflects the previous findings from the Peru Margin, which showed that shallow samples (6.7 and 11 mbsf) tend to cluster in statistical analyses of DGGE banding patterns (Fry et al., 2006) and supports what has been seen in many clone library studies, that the composition of clones changes with depth (Inagaki et al., 2006; Sørensen and Teske, 2006; Nunoura et al., 2009). As already seen from 16S clone library studies, the Chloroflexi group in particular seems to be selected as part of the ‘deep’ community, along with archaeal groups (Teske, 2006; Biddle et al., 2008; Fry et al., 2008). Specific bacterial and archaeal groups are postulated to be well adapted to slow-growing, low-energy lifestyles (Davis et al., 2005; Valentine, 2007). Exactly how these groups achieve their long-term survival remains to be seen, but this thought leads to the suggestion that renewed efforts at cultivating deep phylotypes need to be made. The congruence in taxonomic profiles resulting from these initial PCR-based studies and metagenomic surveys is impressive, given the only partially explored methodological biases of these methods, and the far-reaching implications of primer choice (Teske and Sorensen, 2008).
Also interesting is the apparent association of COG categories from the existing subsurface metagenomes, including that of the Soudan Mine terrestrial system (Figure 5). The comparison of COG categories was restricted to only pyrosequenced samples, to allow for whatever biases that might exist in this analysis to be consistent. Although the Soudan Mine samples looked different in their original publication (Edwards et al., 2006), our reanalysis in light of a much larger dataset shows that they are quite comparable to each other and more similar in their COG structure to shallow sediment samples. Using the lean and obese human gut microbiome as an outgroup, the subsurface metagenomes show that the functional aspect of shallow and terrestrial metagenomes may be more similar to each other than to the deeper Peru Margin metagenomes, which are all very similar to each other. Whether this is due to the actual processes in the sediments allowed by the in-situ geochemistry, or due to the COG profiles of microbial groups that can survive these brutal conditions cannot be determined as of yet. The suggestion that a buried biosphere, regardless of marine or terrestrial location, is exciting and further investigations into deep biosphere environments and comparisons with surface and animal environments will need to be performed to determine if there is a distinct signature of subsurface life.
References
Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH . (2008). Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc Natl Acad Sci USA 105: 10583–10588.
Biddle JF, Lipp JS, Lever MA, Lloyd KG, Sorensen KB, Anderson R et al. (2006). Heterotrophic Archaea dominate the sedimentary subsurface ecosystem off Peru. Proc Natl Acad Sci USA 103: 3846–3851.
Ciccarelli FD, Doerks T, vonMering C, Creevey CJ, Snel B, Bork P . (2006). Toward automatic reconstruction of a highly resolved tree of life. Science 311: 1283–1287.
D’Hondt S, Jorgensen B, Miller DJ, Batzke A, Blake R, Cragg BA et al. (2004). Distributions of microbial activities in deep subseafloor sediments. Science 306: 2216–2221.
D’Hondt S, Spivack AJ, Pockalny R, Ferdelman TG, Fischer JP, Kallmeyer J et al. (2009). Subseafloor sedimentary life in the South Pacific gyre. Proc Natl Acad Sci USA 106: 11651–11656.
Davis KER, Joseph SJ, Janssen PH . (2005). Effects of growth medium, inoculum's size and incubation time on culturability and isolation of soil bacteria. Appl Environ Microbiol 71: 826–834.
Durbin AM, Teske A . (2010). Sediment-associated microdiversity with the marine group i crenarchaeota. Environ Micro Rep 2: 628–705.
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM et al. (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7: 57.
Expedition 308 Scientists (2005). Overpressure and fluid flow processes in the deepwater Gulf of Mexico: slope stability, seeps, and shallow-water flow. IODP Prelim Rep 308: 16–19.
Fry JC, Parkes RJ, Cragg BA, Weightmann AJ, Webster G . (2008). Prokaryotic biodiversity and activity in the deep seafloor biosphere. FEMS Microb Ecol 66: 181–196.
Fry JC, Webster G, Cragg BA, Weightman AJ, Parkes RJ . (2006). Analysis of DGGE profiles to explore the relationship between prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microb Ecol 58: 86–98.
Hugenholtz P, Tyson GW . (2008). Microbiology: metagenomics. Nature 455: 481–483.
Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A et al. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc Natl Acad Sci USA 103: 2815–2820.
Lipp JS, Morono Y, Inagaki F, Hinrichs KU . (2008). Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 454: 991–994.
Meister P, Prokopenko M, Skilbeck CG, Watson M, McKenzie JA . (2005). Data report: Compilation of total organic and inorganic carbon data from Peru Margin and eastern equatorial Pacific drill sites (ODP legs 112, 138, and 201). In Jørgensen BB, D’Hondt S, Miller DJ (eds). Proceedings of the Ocean Drilling Program: Scientific Results, Vol. 201. Available from http://www-odp.tamu.edu/publications/201_SR/VOLUME/CHAPTERS/105.PDF (cited 4 July 2010).
Nunoura T, Soffientino B, Blazekjak A, Kauta J, Oida H, Schippers A et al. (2009). Subseafloor microbial communities associated with rapid turbidite deposition in the Gulf of Mexico continental slope (IODP Expedition 308) FEMS Micro. Ecol 69: 410–424.
Parkes RJ, Webster G, Cragg BA, Weightman AJ, Newberry CJ, Ferdelman TG et al. (2005). Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436: 390–394.
Schippers A, Neretin LN, Kallmeyer J, Ferdelman TJ, Cragg BA, Parkes RJ et al. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433: 861–864.
Sørensen KB, Lauer A, Teske A . (2004). Archaeal phylotypes in a metal-rich, low-activity deep subsurface sediment of the Peru Basin, ODP Leg 201, Site 1231. Geobiology 2: 151–161.
Sørensen KB, Teske A . (2006). Stratified communities of active archaea in deep marine subsurface sediments. Appl Environ Microbiol 72: 4596–4603.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4: 41.
Teske A . (2006). Microbial communities of deep marine subsurface sediments: molecular and cultivation surveys. Geomicrobiol J 23: 357–368.
Teske A, Biddle JF . (2008). Analysis of deep subsurface microbial communities by functional genes and genomics. In: Dilek Y, Furnes H, Muehlenbachs K (eds). Links Between Geological Processes, Microbial Activities & Evolution of Life. Springer, Netherlands.
Teske A, Sorensen K . (2008). Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J 2: 3–18.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2008). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Valentine D . (2007). Adaptations to energy stress dictate the ecology and evolution of Archaea. Nat Rev Microbiol 5: 316–323.
Von Mering C, Hugenholtz P, Raes J, Tringe SG, Doerks T, Jensen LJ et al. (2007). Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 315: 1126–1130.
White JR, Nagarajan N, Pop M . (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5: e1000352.
Whitman WB, Coleman DC, Wiebe WJ . (1998). Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95: 6578–6583.
Acknowledgements
We are indebted to the great efforts of the technical staff on the Joides Resolution during Expedition 308 and the IODP Expedition 308 Scientists, in particular to the shipboard microbiologist T Nunoura. Samples for this research were provided by the Integrated Ocean Drilling Program. The IODP is sponsored by the US National Science Foundation (NSF) and participating countries. We would also like to thank S Schuster and L Tomsho for assistance with sequencing. The GSFLX facility at the Pennsylvania State University Center for Genome Analysis is funded, in part, by a grant from the Pennsylvania Department of Health using Tobacco Settlement Funds appropriated by the legislature. This work was supported by NSF OCE05–5060, the National Aeronautics and Space Administration (NASA) Astrobiology Institute and JOI-USSAC IODP Expedition 308 post-cruise support (CHH). JFB was supported by a NASA Astrobiology Institute Postdoctoral Fellowship administered by Oak Ridge Associated Universities. AT was supported by NAI grant NCC 2-1054. JRW was supported in part by a grant from the Bill and Melinda Gates Foundation (PI: Jim Nataro, subcontract to Mihai Pop).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Biddle, J., White, J., Teske, A. et al. Metagenomics of the subsurface Brazos-Trinity Basin (IODP site 1320): comparison with other sediment and pyrosequenced metagenomes. ISME J 5, 1038–1047 (2011). https://doi.org/10.1038/ismej.2010.199
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2010.199
Keywords
This article is cited by
-
The Microbiome and Urolithiasis: Current Advancements and Future Challenges
Current Urology Reports (2022)
-
Subseafloor life and its biogeochemical impacts
Nature Communications (2019)
-
Unique community structure of viruses in a glacier soil of the Tianshan Mountains, China
Journal of Soils and Sediments (2017)
-
Evidence for extensive gene flow and Thermotoga subpopulations in subsurface and marine environments
The ISME Journal (2015)
-
Single cell genomic study of Dehalococcoidetes species from deep-sea sediments of the Peruvian Margin
The ISME Journal (2014)
