
Abstract
The activities of complex communities of microbes affect biogeochemical transformations in natural, managed and engineered ecosystems. Meaningfully defining what constitutes a community of interacting microbial populations is not trivial, but is important for rigorous progress in the field. Important elements of research in microbial community ecology include the analysis of functional pathways for nutrient resource and energy flows, mechanistic understanding of interactions between microbial populations and their environment, and the emergent properties of the complex community. Some emergent properties mirror those analyzed by community ecologists who study plants and animals: biological diversity, functional redundancy and system stability. However, because microbes possess mechanisms for the horizontal transfer of genetic information, the metagenome may also be considered as a community property.
Similar content being viewed by others


Introduction
Analyses of 16S rRNA gene sequences from environmental DNA have shown the extraordinary richness of phylogenetic types found in many microbial habitats (Ley et al., 2006; Walker and Pace, 2007). As microbial ecologists have learned more about ‘who's there,’ it has heightened interest in understanding ‘what are they doing.’ The concerted activity of interacting microbes was critical to the development of environmental conditions on Earth that led to the evolution of multicellular organisms, and their catalysis of biogeochemical reactions has a central function in sustaining conditions that are compatible with a robust and diverse biosphere. At a time in which human beings are concerned with historically rapid global change, understanding the control mechanisms whereby microbial communities determine ecosystem function is particularly relevant. Current ecosystem simulation models do not include microbial composition, and often neither explicitly consider the effects of environmental conditions on microbial activities nor the interactions between diverse microbial processes (Bardgett et al., 2008). As microbial communities are of primary importance in biogeochemical transformations, a deeper understanding of their dynamics will be critical to refined predictions regarding how the biosphere modulates and responds to future environmental conditions. At a more fundamental level, understanding natural microbial communities will deepen our understanding of how ecosystems function. In turn, this understanding may elucidate novel interaction mechanisms among multiple species at the single cell level (O’Malley and Dupre, 2007).
There is great interest in applying modern technologies in genome-enabled biology and analytical chemistry to concurrently study both the identity and the function of complex microbial communities (DeLong et al., 2006; Musat et al., 2008). However, effective application requires a clear delineation of what is meant by a ‘microbial community’ and identification of important characteristics specific to community ecology.
What is a microbial community?
The concept of community ecology arose in plant and animal ecology. Communities are defined as multi-species assemblages, in which organisms live together in a contiguous environment and interact with each other. This discipline seeks to analyze how biological assemblages are structured, what are their functional interactions and how community structure changes in space and time. Clements (1916) viewed the community as a ‘supra-organism’, which had a well-defined level of organization with tight interactions among organisms that comprise a causal system and gives rise to emergent properties. The boundaries of the community may have less to do with physical dimensions than the range over which there are strong rather than weak interactions between populations (Levins and Lewontin, 1985). The alternative individualistic concept (Gleason, 1926) is that many species co-occur in a habitat because they tolerate similar physical and chemical conditions and do not necessarily interact with each other. The practical delineation of ‘community’ may then reflect the interests of the ecologist rather than any inherent characteristics.
The problems in rigorously defining community are heightened in the case of microbial ecology. In particular, delineating a ‘contiguous environment’ and the meaning of ‘interact’ may be problematic. Microorganisms react to and in turn influence conditions in their microenvironments, which usually have length scales of microns rather than millimeters (Young et al., 2008), except in cases of multicellular structures such as fungal hyphae. However, the consumption of substrates and production of metabolic products in water-saturated sediments and density-stabilized aquatic water columns can generate chemical gradients over meters (Wakeham et al., 2007). The consequence is that these functional groups metabolically ‘interact’ over many meters. As a result, the strength of interaction among organisms and the defined spatial scale may vary substantially for investigator-defined microbial communities.
To increase rigor in the meaning of ‘microbial community,’ it would be valuable for microbial ecologists to explicitly articulate their meaning for each specific research effort. Microbes strongly interacting with each other in a microenvironment comprise a local community. However, the distribution of organisms and physicochemical properties within most habitats is patchy; even in apparently well-mixed oligotrophic planktonic habitats, nutrient-rich foci of marine snow may occur (Azam and Malfatti, 2007). The patchwork of local communities has been termed as a phenomenological community (Sterelny, 2006); for the purposes of microbial ecology, this would represent a range of macroscale habitats delineated by the investigator, in which the assemblage of microbes persists in spatial association. The phenomenological community could be constrained to a smaller number of populations by defining an indexical community—the set of populations that directly interact with a key population or defined biogeochemical process, together with other local populations that affect the directly interacting populations (Sterelny, 2006).
Recent developments in community ecology have begun to recognize that the biological assemblage cannot be defined without reference to its abiotic environment. An appreciation for the tight interrelationship between microbes and their microscale physical and chemical environments is particularly important for delineation of microbial communities (O’Donnell et al., 2007). In this spirit, it may be instructive to define microbial communities not from a macroscale perspective (for example aquatic vs terrestrial habitats), but rather based on a bottom-up analysis of the physicochemical characteristics of the microenvironment, with upscaling to a spatial domain (the ‘contiguous environment’) defined by the region over which substantial direct interactions or indirect chemical interactions are occurring. This approach presupposes adequate analysis of the local physicochemical environment, but technical innovations are moving to the microscale level (Young et al., 2008). Konopka (2006) defined four ecosystems derived by considering the environment from the microbe's local perspective (Table 1). Each has particular characteristics that define important selective forces in that habitat, but which also impact the spatial scale over which microbial interactions occur.
Elements of microbial community analysis
Beyond the rigorous definition of a microbial community, there remains the elucidation of its essential characteristics. Levin (1999) proposed that ‘the most important challenge for ecologists remains to understand the linkages between what is going on at the level of the physiology and behavior of individual organisms and emergent properties such as the productivity and resiliency of ecosystems.’ From this perspective, important elements for analysis of microbial communities comprise fluxes, interactions and properties.
Analysis of community functional pathways
Microbial communities provide ecosystem services (Ducklow, 2007) through the catalysis of biogeochemical reactions. Many reactions in chemoheterotrophs entail conversion of chemical elements from organic forms to inorganic forms that can be used by primary producers for new biomass synthesis. Others interconvert elements between oxidized and reduced forms, and these coupled reactions can sustain the interacting organisms and have great impact on geological processes (Ehrlich, 1998). Lists of these reactions can be found in the description of biogeochemical cycles (Fenchel et al., 1998).
The analysis of material and energy flow through distributed metabolic networks of microbes could lead to a predictive understanding of ecosystems (Roling et al., 2007). However, it requires a deeper and richer analysis than is presently available. For example, models of terrestrial carbon cycling posit only a few pools of organic C that differ in first-order decay rates (Causarano et al., 2007). Soil organic matter consists of hundreds of different organic substrates, heterogeneously distributed in a three-dimensional matrix. Fifty thousand different microbial taxa may be present in a kilogram of soil (Roesch et al., 2007). Technically, it is possible to measure the rates of specific biogeochemical processes and to census the phylogenetic identities of the resident microbes. It is more difficult to determine which taxon is responsible for a specific biogeochemical process in situ (Madsen, 2005). However, increasingly sophisticated analytical methods such as NanoSIMS and Raman spectroscopy can permit the use of stable isotopes to measure process rates, with subsequent analyses of labeled macromolecules to phylogenetically identify the microbial catalyst (Behrens et al., 2008; Huang et al., 2009).
Interactions among organisms
Interest in microbial community ecology is a consequence of the postulate that interactions between organisms (microbe–microbe and microbe–metazoan) are of essential importance for understanding ecosystem dynamics and the evolutionary ecology of individual organisms. However, this remains an area in which the cataloging of potential interactions is better developed than mechanistic understanding of specific cases. The field of microbial ecology has correctly been called to task for an insufficient body of theory (Prosser et al., 2007). However, this is one area in which theories on the effects of interactions on community diversity (Reichenbach et al., 2007; Beninca et al., 2008) exceed detailed experimental verification.
Although interspecies competition for nutrient resources is presumed to be widespread, it is difficult to analyze in natural communities; more typically, one detects its outcome as the composition of the microbial community. Laboratory studies of simple, constructed communities and theoretical investigations can provide insights into important molecular mechanisms (Gottschal, 1993; Prado and Kerr, 2008). Interactions can reflect cytoplasmic or extracytoplasmic activities, such as the affinity of membrane-bound transporters for limiting substrates (Robin et al., 2008) or the production of cell-surface-associated or extracellular enzymes for macromolecule hydrolysis (Allison, 2005). However, even at our current state of knowledge, a catalog of microbial interactions (Table 2) goes far beyond simple resource competition (Little et al., 2008). These include not only short-term (physiological) interactions, but also potentially deeper consequences that arise from horizontal gene transfer or co-evolution of traits between different microbes or a microbe and a multicellular organism. Although we have ‘textbook’ examples of these phenomena, the breadth of their ecological significance awaits discovery.
Emergent properties
Emergent properties of a community are characteristics not identifiable by analyzing the component organisms in isolation. Properties of this type can arise when the system is viewed at a high level of organization. An analysis of these properties can lead to an understanding of how the system responds to both external and internal perturbations, and provides the capability to predict system dynamics in response to environmental change.
Taxonomic diversity
The central property in community ecology is diversity. An inventory of diversity within a discrete sample has two important components—taxon richness and the relative abundance of different taxa in the community. The analysis of 16S rRNA gene sequences directly from environmental samples (Tringe and Hugenholtz, 2008) has greatly enriched our understanding of global microbial diversity. However, the financial costs of sequencing in the past meant that habitats were usually grossly undersampled relative to their richness. However, new sequencing technologies can now remove that restriction, and even very rare taxa can be detected (Sogin et al., 2006). One benefit of deeper sampling is a more precise determination of the relative abundance of different taxa in a habitat. Analysis of relative abundance is conceptually very important to the other properties of communities discussed below, but a consequence of undersampling is that detection and quantitation of taxa present at low relative abundance is imprecise. New DNA sequencing technologies (pyrosequencing) can provide enough data (although more limited phylogenetic resolution), and the application of theoretical constructs to community assembly (Curtis et al., 2006) can suggest methods of analysis that provide informative rank-abundance distributions for microbial communities.
The term ‘diversity’ can be applied at different ecological scales, and it is important to be clear about the intended meaning. Whittaker (summarized in Magurran, 2004) described a matrix of diversity indices that can be applied to microbial systems at a wide range of spatial scales. Complementary information is provided by inventory diversity (taxonomic diversity within spatially defined habitats) and differentiation diversity indices (the variation in an inventory diversity index). Microbial ecologists generally fail to accurately articulate which diversity index they have measured. It is important to understand whether those analyses represent differentiation of pattern diversity (as would be true of a large number of subsamples from local environments) or measures of between-habitat diversity.
Functional diversity and functional redundancy
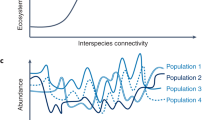
The analysis of taxonomic diversity across the microbial world is important on its own, as it illuminates the richness of evolutionary history over the past 3 billion years. However, for the analysis of microbial community ecology and the functioning of ecosystems, the determination of functional diversity is of greatest importance. Higher levels of redundancy for ecological functions should be related to the reliability with which an ecosystem will continue to deliver services in the face of environmental changes (Naeem et al., 1998) (Figure 1). The central question is how to assay and quantify functional diversity. Phylogeny is informative to an adequate level in a small number of cases (for example methane-oxidizing bacteria (McDonald et al., 2008)). However, it is more common to find that specific ecological functions are distributed across a wide breadth of taxa (nitrogen fixation and denitrification provide two examples).

Conceptual model of relationship between functional redundancy and ecosystem stability (its resistance and resiliency to environmental perturbations). As new taxa are added to a pristine ecosystem, the number of ecological functions that are present increases. However, at some point, additional taxa do not add novel capacities, but do add redundancy to specific functions. Highly redundant systems are more likely to retain a function after an environmental stress because the probability of a resident taxon that is stress resistant is greater.
In some cases, there are conserved regions of functional genes (such as sulfite reductase dsrA for sulfate reduction (Miletto et al., 2007) and nitrite or nitrous oxide reductases for nitrate reduction (Mills et al., 2008)) that can be used to assess the abundance of these genes in natural samples. Analysis of sequence differences among amplicons by fingerprinting techniques (Wertz et al., 2006) will permit an analysis of functional diversity, at least for abundant community members. On the other hand, there are other important functions such as extracellular electron transfer, in which key functional proteins, extracytoplasmic multiheme cytochromes, found in metal-reducing bacteria such as Anaeromyxobacter, Geobacter and Shewanella species, are poorly conserved. The other limitation to this molecular approach for analysis of functional diversity is that novel genes and enzymes will not be recognized.
A weakness of most current applications of community metagenomics or metaproteomics to complex communities is that gene sequences and proteins are disassociated from the intact organism that possessed them. As a result, the analyses cannot place a specific function in the context of the other functional properties of the organism. A measure of functional redundancy could be derived from cataloging genes or proteins with annotated functions and correlated to ecosystem resiliency to experimentally imposed perturbations (such as Hg stress). However, a mechanistic understanding will require analysis of the co-occurrence of those functional genes or gene products with stress resistance determinants in discrete organisms, either by bioinformatic analysis of single genomes or physiological analyses of cultivated organisms. For complex microbial communities, we may have to wait for the advent of technologies, whereby several hundred bacterial cells could be individually plucked from a habitat, and their genomes amplified (if necessary) and sequenced in a high-throughput and inexpensive manner. In this way, the community metagenome would comprise a list of functional genes in the complete context of the other genetic determinants within the organism.
Stability/resistance/resilience
Each of these terms has been applied to a description of ecosystem response to environmental perturbations. Resistance is related to the degree that a system does not exhibit an acute loss of function after imposition of a stress, whereas resilience connotes the rate at which functionality returns after an acute stress effect (Pimm, 1984). ‘Stability’ could then represent either the resistance of a system to acute, toxic effects or resilience, taking into account a longer-term perspective, in which stress imposes selection for resistant taxa present at low relative abundance. The stability concept refers to the functional properties of the community (Tilman, 1999), rather than to the population sizes of the constituents, as the latter are very dynamic in response to environmental changes.
Theoretical and empirical work in ecology has investigated the idea that higher diversity within a community will lead to increased ecosystem stability (Loreau, 2000; Tilman et al., 2006). Several mechanisms have been put forward for this; these mechanisms are not mutually exclusive: (i) individual ecotypes may differ in their effects on ecosystem processes; thus, particular combinations of ecotypes may result in distinctive ecosystem properties. However, layered on top of this are empirical observations of (ii) a richness effect, in which a mixture of ecotypes performs differently than the average performance of the individuals in isolation. There are many possible mechanisms, and microbial ecology represents an experimentally tractable discipline, in which it may be possible to identify the molecular mechanisms. Richness effects can arise from complementary properties of taxa within the communities such as niche portioning or facilitation (mutualistic or commensal interactions). Alternatively, some taxa may have dominant effects and there is a higher probability that one of these dominant forms is present in a highly diverse community (the sampling effect).
Community genetic potential
The community properties articulated above have all been developed within plant and animal ecology and are applicable to microbial community ecology. However, it is useful to consider whether microbial communities possess a unique community property—the community metagenome. Microbes (particularly Bacteria and Archaea) have mechanisms for gene transfer across broad phylogenetic barriers that do not occur at ecologically significant rates in other organisms. Thus, the resilience of microbial communities over a period of a few years to environmental perturbations may rest in part on the frequency of horizontal gene transfer within microbial communities. Two examples of responsiveness under strong selection pressure are the dissemination of antibiotic resistance determinants (Hughes and Datta, 1983) and the development of metabolic pathways for the catabolism of novel xenobiotic organic compounds (Juhas et al., 2009).
This suggests a role for the cataloging of genes (metagenomes) or protein sequences (metaproteomes) from microbial communities. The technology exists to conduct these surveys. However, substantial advances in informatics are still required to make them useful in an ecological sense. The specificity of annotation that is satisfactory for comparative genomics is inadequate to define ecological functionality. That is, analysis of microbial community functionality requires not only the identification of a protein family, but also an understanding of the range of substrate usage. A larger problem is the substantial fraction (approximately 30%) of genes in sequenced bacterial genomes, for which no function is assignable at all. Perhaps their functions are unknown because they are unimportant under laboratory culture, which may mean that they are very significant for some ecological function in nature, but this would require significant experimentation to uncover.
In summary, molecular censuses of microbes in nature have uncovered a breathtaking genetic diversity; in addition, new biogeochemical pathways and catalysts continue to be discovered by cultivation, albeit at a slower pace. In the past few years, technologies that sequence DNA, mRNA or proteins have been applied to natural communities as ‘discovery-based’ science. This is appropriate at an early phase to search for patterns (now at the level of genes and proteins rather than taxa) that can lead to hypotheses regarding the causes of those patterns. However, one route to produce functional connections between microbial diversity and ecosystem properties is the rigorous application of community ecology concepts to microbial ecosystems, in ways that are testable and lead to a predictive synthesis. Some of the technical opportunities and challenges for future research were mentioned above: the increased sensitivity of stable isotope analyses through new mass spectrometry techniques that can be coupled to phylogenetic analyses of labeled macromolecules to identify the important microbial catalysts and the major challenge of extracting precise functional information from the annotation of nucleic acid or protein sequences.
Additional directions for future research that would aid in going beyond a descriptive to a predictive science of microbial community ecology might include several other experimental, computational and technical approaches:
-
1)
Expansion of experimental approaches beyond those most commonly used in microbial ecology: a common experimental regime is manipulation of environmental factors in the field or in confined samples to analyze effects of abiotic factors on ecosystem processes. However, it is difficult to assess how microbial community structure impacts ecosystem functioning from these experiments. Application of experimental approaches such as common garden experiments and reciprocal transplants can provide community-level insights (Reed and Martiny, 2007).
-
2)
Predictive models at the community scale: this is a daunting task, and current models of aquatic or terrestrial ecosystems (Izaurralde et al., 2006) neither attempt to include microbiological diversity nor spatial heterogeneities. A path forward would include scaling at the fundamental microbial ecosystem level (Table 1). For example, in terrestrial ecosystems, individual soil aggregates could represent an appropriate scale of community structure; one can collect data on the physical structure of the matrix, and modeling techniques (such as individual-based approaches) can be applied to incorporate biological diversity and spatial heterogeneity (O’Donnell et al., 2007). This individual-based approach has been applied to multi-species biofilms (Picioreanu et al., 2004). If these modeling approaches were successful, other computational techniques would be necessary to upscale predictions to larger spatial scales.
-
3)
Microbial model systems to test ecological theory: microcosm experiments have been criticized as unrepresentative surrogates of natural ecosystems (see Jessup et al., 2004). However, the perceived constraints of these experiments also provide the means to rigorously test sophisticated ecological theories—the experimentalist can simplify a system to its fundamentals, control the scale and effects of environmental factors and reduce spatial heterogeneity. A recent example (Wittebolle et al., 2009) illustrated how >1000 microbial microcosms were deployed to test the effects of evenness in relative abundance on functional stability of ecosystems. The positive effects of evenness on the constructed microbial communities’ responses to environmental stressors have implications for larger-scale ecosystems in which invasive species or anthropogenic activities have altered community structure (Naeem, 2009).
-
4)
Explicitly directing research efforts to the three specific elements of microbial community analysis identified above: the analysis of community functional pathways will benefit from the increasing sophistication of stable isotope analyses mentioned above; a limitation is that instrumentation is currently rare because of its expense. There are also new, mass spectrometric-based methods to resolve the complex suite of organic substrates present in soil or water, although the determination of molecular formulas from these data is in its infancy (Reemtsma, 2009). Among the emergent properties of microbial communities, functional redundancy is one that is of substantial theoretical interest, and which might be tractable to measure, at least for some well-understood biogeochemical functions (examples include nitrogen fixation and aromatic dioxygenases). This can be accomplished through data mining of metagenomes or targeted approaches such as PCR amplification of known functional genes or development of activity-based proteomic approaches (Cravatt et al., 2008).
References
Allison SD . (2005). Cheaters, diffusion and nutrients constrain decomposition by microbial enzymes in spatially structured environments. Ecol Lett 8: 626–635.
Azam F, Malfatti F . (2007). Microbial structuring of marine ecosystems. Nat Rev Microbiol 5: 782–791.
Bardgett RD, Freeman C, Ostle NJ . (2008). Microbial contributions to climate change through carbon cycle feedbacks. ISME J 2: 805–814.
Behrens S, Losekann T, Pett-Ridge J, Weber PK, Ng WO, Stevenson BS et al. (2008). Linking microbial phylogeny to metabolic activity at the single-cell level by using enhanced element labeling-catalyzed reporter deposition fluorescence in situ hybridization (EL-FISH) and NanoSIMS. Appl Environ Microbiol 74: 3143–3150.
Beninca E, Huisman J, Heerkloss R, Johnk KD, Branco P, Van Nes EH et al. (2008). Chaos in a long-term experiment with a plankton community. Nature 451: 822–825.
Bertin C, Yang XH, Weston LA . (2003). The role of root exudates and allelochemicals in the rhizosphere. Plant Soil 256: 67–83.
Causarano HJ, Shaw JN, Franzluebbers AJ, Reeves DW, Raper RL, Balkcom KS et al. (2007). Simulating field-scale soil organic carbon dynamics using EPIC. Soil Sci Soc Am J 71: 1174–1185.
Clements FE . (1916). Plant Succession: An Analysis of the Development of Vegetation. Carnegie Institution: Washington, D.C.
Cooper JE . (2007). Early interactions between legumes and rhizobia: disclosing complexity in a molecular dialogue. J Appl Microbiol 103: 1355–1365.
Cravatt BF, Wright AT, Kozarich JW . (2008). Activity-based protein profiling: from enzyme chemistry. Annu Rev Biochem 77: 383–414.
Curtis TP, Head IM, Lunn M, Woodcock S, Schloss PD, Sloan WT . (2006). What is the extent of prokaryotic diversity? Philos Trans R Soc B Biol Sci 361: 2023–2037.
DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, Frigaard NU et al. (2006). Community genomics among stratified microbial assemblages in the ocean's interior. Science 311: 496–503.
Des Marais DJ . (2003). Biogeochemistry of hypersaline microbial mats illustrates the dynamics of modern microbial ecosystems and the early evolution of the biosphere. Biol Bull 204: 160–167.
Dolfing J . (2001). The microbial logic behind the prevalence of incomplete oxidation of organic compounds by acetogenic bacteria in methanogenic environments. Microb Ecol 41: 83–89.
Ducklow H . (2007). 10th Symposium on Aquatic Microbial Ecology (SAME 10). Faro: Portugal, pp 13–19.
Ehrlich HL . (1998). Geomicrobiology: its significance for geology. Earth-Sci Rev 45: 45–60.
Fenchel T, King G.M, Blackburn H . (1998). Bacterial Biogeochemistry: The Ecophysiology of Mineral Cycling. Academic Press: San Diego.
Gillor O, Etzion A, Riley MA . (2008). The dual role of bacteriocins as anti- and probiotics. Appl Microbiol Biotechnol 81: 591–606.
Gleason HA . (1926). The Individualistic Concept of the Plant Association. Bull Torrey Botanical Club 53: 7–26.
Gonzalez JE, Keshavan ND . (2006). Messing with bacterial quorum sensing. Microbiol Molec Biol Rev 70: 859–875.
Gottschal JC . (1993). Growth-kinetics and competition—some contemporary comments. Antonie Van Leeuwenhoek Int J Gen Mol Microbiol 63: 299–313.
Huang WE, Ferguson A, Singer AC, Lawson K, Thompson IP, Kalin RM et al. (2009). Resolving genetic functions within microbial populations: in situ analyses using rRNA and mRNA stable isotope probing coupled with single-cell Raman-fluorescence in situ hybridization. Appl Environ Microbiol 75: 234–241.
Hughes VM, Datta N . (1983). Conjugative plasmids in bacteria of the pre-antibiotic era. Nature 302: 725–726.
Izaurralde RC, Williams JR, McGill WB, Rosenberg NJ, Jakas MCQ . (2006). Simulating soil C dynamics with EPIC: model description and testing against long-term data. Ecol Modell 192: 362–384.
Jessup CM, Kassen R, Forde SE, Kerr B, Buckling A, Rainey PB et al. (2004). Big questions, small worlds: microbial model systems in ecology. Trends Ecol Evol 19: 189–197.
Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW . (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33: 376–393.
Konopka A . (2006). Microbial ecology: searching for principles. Microbe 1: 175–179.
Levin S . (1999). Fragile Dominion: Complexity and the Commons. Basic Books: New York.
Levins R, Lewontin RC . (1985). Dialectics and reduction in ecology. In: Levins R and Richard C Lewontin (eds). The Dialectical Biologist. Harvard University Press: Cambridge, pp 132–160.
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM et al. (2006). Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72: 3685–3695.
Little AEF, Robinson CJ, Peterson SB, Raffa KE, Handelsman J . (2008). Rules of engagement: interspecies interactions that regulate microbial communities. Annu Rev Microbiol 62: 375–401.
Loreau M . (2000). Biodiversity and ecosystem functioning: recent theoretical advances. Oikos 91: 3–17.
Madsen EL . (2005). Identifying microorganisms responsible for ecologically significant biogeochemical processes. Nat Rev Microbiol 3: 439–446.
Magurran AE . (2004). Measuring Biological Diversity. Blackwell Science, Oxford, UK.
McDonald IR, Bodrossy L, Chen Y, Murrell JC . (2008). Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol 74: 1305–1315.
Miethke M, Marahiel MA . (2007). Siderophore-based iron acquisition and pathogen control. Microbiol Molec Biol Rev 71: 413–451.
Miletto M, Bodelier PLE, Laanbroek HJ . (2007). Improved PCR-DGGE for high resolution diversity screening of complex sulfate-reducing prokaryotic communities in soils and sediments. J Microbiol Methods 70: 103–111.
Mills HJ, Hunter E, Humphrys M, Kerkhof L, McGuinness L, Huettel M et al. (2008). Characterization of nitrifying, denitrifying, and overall bacterial communities in permeable marine sediments of the northeastern Gulf of Mexico. Appl Environ Microbiol 74: 4440–4453.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and Evolution of Heritable Bacterial Symbionts. Annu Rev Genet 42: 165–190.
Musat N, Halm H, Winterholler B, Hoppe P, Peduzzi S, Hillion F et al. (2008). A single-cell view on the ecophysiology of anaerobic phototrophic bacteria. Proc Natl Acad Sci USA 105: 17861–17866.
Naeem S . (2009). Ecology Gini in the bottle. Nature 458: 579–580.
Naeem S, Kawabata Z, Loreau M . (1998). Transcending boundaries in biodiversity research. Trends Ecol Evol 13: 134–135.
O’Donnell AG, Young IM, Rushton SP, Shirley MD, Crawford JW . (2007). Visualization, modelling and prediction in soil microbiology. Nat Rev Microbiol 5: 689–699.
O’Malley MA, Dupre J . (2007). Size doesn’t matter: towards a more inclusive philosophy of biology. Biol Philos 22: 155–191.
Overmann J, van Gemerden H . (2000). Microbial interactions involving sulfur bacteria: implications for the ecology and evolution of bacterial communities. FEMS Microbiol Rev 24: 591–599.
Picioreanu C, Kreft JU, van Loosdrecht MCM . (2004). Particle-based multidimensional multispecies biofilm model. Appl Environ Microbiol 70: 3024–3040.
Pimm SL . (1984). The complexity and stability of ecosystems. Nature 307: 321–326.
Prado F, Kerr B . (2008). The evolution of restraint in bacterial biofilms under nontransitive competition. Evolution 62: 538–548.
Prosser JI, Bohannan BJM, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP et al. (2007). Essay—the role of ecological theory in microbial ecology. Nat Rev Microbiol 5: 384–392.
Reed HE, Martiny JBH . (2007). Testing the functional significance of microbial composition in natural communities. FEMS Microbiol Ecol 62: 161–170.
Reemtsma T . (2009). Determination of molecular formulas of natural organic matter molecules by (ultra-) high-resolution mass spectrometry status and needs. J Chromatogr A 1216: 3687–3701.
Reichenbach T, Mobilia M, Frey E . (2007). Mobility promotes and jeopardizes biodiversity in rock-paper-scissors games. Nature 448: 1046–1049.
Riley MA, Wertz JE . (2002). Bacteriocins: Evolution, ecology, and application. Annu Rev Microbiol 56: 117–137.
Robin A, Vansuyt G, Hinsinger P, Meyer JM, Briat JF, Lemanceau P . (2008). Iron dynamics in the rhizosphere: consequences for plant health and nutrition. Adv Agronomy 99: 183–225.
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Roling WFM, van Breukelen BM, Bruggeman FJ, Westerhoff HV . (2007). Ecological control analysis: being(s) in control of mass flux and metabolite concentrations in anaerobic degradation processes. Environ Microbiol 9: 500–511.
Ryan RP, Dow JM . (2008). Diffusible signals and interspecies communication in bacteria. Microbiol-SGM 154: 1845–1858.
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Sowell SM, Wilhelm LJ, Norbeck AD, Lipton MS, Nicora CD, Barofsky DF et al. (2009). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J 3: 93–105.
Sterelny K . (2006). Local ecological communities. Philos Sci 73: 215–231.
Suttle CA . (2007). Marine viruses—major players in the global ecosystem. Nat Rev Microbiol 5: 801–812.
Tilman D . (1999). The ecological consequences of changes in biodiversity: a search for general principles. Ecology 80: 1455–1474.
Tilman D, Reich PB, Knops JMH . (2006). Biodiversity and ecosystem stability in a decade-long grassland experiment. Nature 441: 629–632.
Tringe SG, Hugenholtz P . (2008). A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11: 442–446.
Villarreal DT, Turco RF, Konopka A . (1991). Propachlor degradation by a soil bacterial community. Appl Environ Microbiol 57: 2135–2140.
Wakeham SG, Amann R, Freeman KH, Hopmans EC, Jorgensen BB, Putnam IF et al. (2007). Microbial ecology of the stratified water column of the Black Sea as revealed by a comprehensive biomarker study. Org Geochem 38: 2070–2097.
Walker JJ, Pace NR . (2007). Endolithic microbial ecosystems. Annu Rev Microbiol 61: 331–347.
Wanner G, Vogl K, Overmann J . (2008). Ultrastructural characterization of the prokaryotic symbiosis in ‘Chlorochromatium aggregatum’. J Bacteriol 190: 3721–3730.
Wertz S, Degrange V, Prosser JI, Poly F, Commeaux C, Freitag T et al. (2006). Maintenance of soil functioning following erosion of microbial diversity. Environ Microbiol 8: 2162–2169.
Wittebolle L, Marzorati M, Clement L, Balloi A, Daffonchio D, Heylen K et al (2009). Initial community evenness favours functionality under selective stress. Nature 458: 623–626.
Young IM, Crawford JW, Nunan N, Otten W, Spiers A . (2008). Microbial distribution in soils: physics and scaling. Adv Agronomy 100: 81–121.
Acknowledgements
This work was conducted in part under the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory, a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract DE-AC05-76RL01830.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Konopka, A. What is microbial community ecology?. ISME J 3, 1223–1230 (2009). https://doi.org/10.1038/ismej.2009.88
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2009.88
Keywords
This article is cited by
-
VESPA: an optimized protocol for accurate metabarcoding-based characterization of vertebrate eukaryotic endosymbiont and parasite assemblages
Nature Communications (2024)
-
Uncovering the microbiome landscape in sashimi delicacies
Scientific Reports (2024)
-
The Microbiome Function in a Host Organism: A Medical Puzzle or an Essential Ecological Environment?
Biological Theory (2024)
-
High-energy diet modify rumen microbial composition and microbial energy metabolism pattern in fattening sheep
BMC Veterinary Research (2023)
-
Pedogenesis of typical zonal soil drives belowground bacterial communities of arable land in the Northeast China Plain
Scientific Reports (2023)
