
Abstract
The maintenance of a low pH in the vagina through the microbial production of lactic acid is known to be an important defense against infectious disease in reproductive age women. Previous studies have shown that this is largely accomplished through the metabolism of lactic acid bacteria, primarily species of Lactobacillus. Despite the importance of this defense mechanism to women's health, differences in the species composition of vaginal bacterial communities among women have not been well defined, nor is it known if and how these differences might be linked to differences in the risk of infection. In this study, we defined and compared the species composition of vaginal bacterial communities in 144 Caucasian and black women in North America. This was carried out based on the profiles of terminal restriction fragments of 16S rRNA genes, and phylogenetic analysis of 16S rRNA gene sequences of the numerically dominant microbial populations. Among all the women sampled, there were eight major kinds of vaginal communities (‘supergroups’) that occurred in the general populace at a frequency of at least 0.05 (P=0.99). From the distribution of these supergroups among women, it was possible to draw several conclusions. First, there were striking, statistically significant differences (P=0.0) in the rank abundance of community types among women in these racial groups. Second, the incidence of vaginal communities in which lactobacilli were not dominant was higher in black women (33%) as compared to Caucasian women (7%). Communities not dominated by lactobacilli had Atopobium and a diverse array of phylotypes from the order Clostridiales. Third, communities dominated by roughly equal numbers of more than one species of Lactobacillus were rare in black women, but common in Caucasian women. We postulate that because of these differences in composition, not all vaginal communities are equally resilient, and that differences in the vaginal microbiota of Caucasian and black women may at least partly account for known disparities in the susceptibility of women in these racial groups to bacterial vaginosis and sexually transmitted diseases.
Similar content being viewed by others
Introduction
The high levels of estrogen in reproductive age women cause large amounts of glycogen to be deposited in the vaginal epithelium (Paavonen, 1983), and it is subsequently metabolized primarily by bacterial populations to produce organic acids (Boskey et al., 1999). The resulting low pH (4–4.5) of the vagina creates a restrictive environment that precludes the growth of many pathogenic organisms. Over the years, a large number of different species of lactic acid bacteria, primarily species of Lactobacillus, have been demonstrated to reside in the human vagina (Redondo-Lopez et al., 1990; Antonio et al., 1999) and be key players in this process. These species effectively constitute an ecological guild – a group of species that have similar requirements and play a similar role within a community – and maintaining high numbers of these populations is a hallmark of healthy conditions. Events that lead to decreased numbers of lactic acid bacteria in the vagina and the concomitant increase in the abundances of other bacteria are collectively known as bacterial vaginosis (BV; Thorsen et al., 1998; Koumans and Kendrick, 2001). During BV, there is an increase in vaginal pH (>4.5) that is often accompanied by the production of odor, and a thin, grayish white vaginal discharge (Sobel, 2000).
BV commonly occurs in women of reproductive age. It affects between 20% and 25% of the general population, and up to 50% of women attending sexual health clinics (Newton et al., 1997; Morris et al., 2001; Yen et al., 2003; Sobel, 2005). It is the most common cause of vaginal symptoms prompting women to seek medical care (Morris et al., 2001; Marrazzo, 2006), and a significant problem because it predisposes individuals to sexually transmitted diseases including human immunodeficiency virus (HIV) (Sewankambo et al., 1997; Taha et al., 1998; Martin et al., 1999), preterm birth, pelvic inflammatory disease, endometritis and infertility (Hillier et al., 1995; Sweet, 1995; Wiesenfeld et al., 2002; Leitich et al., 2003; Haggerty et al., 2004). The causes of BV are not well understood, and specific infectious agent(s) have not been identified. However, a number of bacterial species are known to be common in women with BV, including Gardnerella vaginalis, Mobiluncus sp., Mycoplasma hominis and species of Clostridiaceae (Thorsen et al., 1998; Koumans and Kendrick, 2001; Fredricks et al., 2005), but their role in the pathogenesis of BV has not been established. The occurrence of BV has been associated with behaviors that upset the normal balance of bacteria in the vagina including new or multiple sex partners, douching, and the use of an intrauterine device for contraception (http://www.cdc.gov/std/bv/STDFact-Bacterial-Vaginosis). This has led some to suggest that BV is not an infectious disease at all but rather a set of symptoms that reflect an ecological disturbance that is accompanied by changes in the relative abundance of autochthonous organisms in the vagina (http://www.cdc.gov/std/bv/STDFact-Bacterial-Vaginosis). Curiously, the incidence of BV varies markedly among racial and ethnic groups (Rajamanoharan et al., 1999; Royce et al., 1999), ranging from 6% in Asians and 9% in whites, to 16% in Hispanics and 23% in African Americans. The reasons for differences in the incidence of BV among racial groups are unknown, but they cannot be explained by differences in socio-demographics, sexual activity, health behavior or hygiene alone (Goldenberg et al., 1996; Royce et al., 1999).
While there are many reasons for concern about BV, the link between BV and an increased risk for acquisition of the HIV (Sewankambo et al., 1997; Taha et al., 1999) is particularly menacing. The number of women with HIV and acquired immunodeficiency syndrome (AIDS) has steadily increased worldwide and by the end of 2005, 17.5 million women were infected with HIV (http://www.niaid.nih.gov/factsheets/womenhiv.htm). More than 90% of HIV infections in adolescent and adult females result from heterosexual intercourse, where the risk of acquiring HIV is 2–4 times higher if the woman has BV (Sewankambo et al., 1997; Martin et al., 1999; Taha et al., 1999). Myer et al. (2005) argue that interventions to reduce the occurrence of BV may have an impact on the spread of HIV, and that almost one-third of all new HIV infections might be prevented if all cases of BV could be cured. However, the existing treatments for BV (oral or vaginal administration of metronidazole) are plagued by high failure rates, with symptoms reoccurring in 54% of women within 3 months following antibiotic treatment (Bradshaw et al., 2006). Measures that stabilize vaginal microbial communities and prophylaxis against BV may emerge if we develop an accurate understanding of the species composition and ecology of vaginal ecosystems in normal healthy women.
In this study, we tested the hypothesis that there were fundamental differences in the composition and structure of bacterial communities in the vaginas of Caucasian and black women in North America. To do this, we determined the identity and rank abundance of bacterial populations in vaginal bacterial communities of 144 healthy, reproductive age women in these racial groups. The data showed that within each racial group there were distinct differences in community composition, which allowed us to group women into categories that we referred to as ‘supergroups’. There were significant differences in the rank abundance of these communities in Caucasian and black women, and we postulate that these differences in community composition and structure may account for the differential risk to BV and various vaginal infections of women in these racial groups.
Materials and methods
Subjects and clinical study design
As part of a study on the prevalence of Staphylococcus aureus carriage, 3012 healthy menstruating women between the ages of 13 and 40 years were enrolled from five sites in North America (Parsonnet et al., 2005). The subjects were recruited by Hill Top Research Inc. in Cincinnati, OH, USA, East Brunswick, NJ, USA, St Petersburg, FL, USA, Scottsdale, AZ, USA and Winnipeg, Manitoba, Canada. The racial profile of the women corresponded with that of the 1990 USA census: 80% white, 12% black, 5% Hispanic and 3% Asian. Subjects were eligible for enrollment if they had regular menstrual cycles (21–35 days); used tampons at least occasionally; were able to read, write and understand English; did not bathe or shower within 2 h of their scheduled visit; refrained from douching, vaginal medications, suppositories, feminine sprays, genital wipes or contraceptive spermicides for 48 h before their scheduled visit; and were willing to comply with all other protocol requirements. Subjects were not eligible if they were participating in another clinical study; were pregnant, actively trying to get pregnant or suspected they were pregnant; had a gynecological abnormality as judged by the study's medical personnel; had an infection of the genitals within the past 6 weeks; had been medically diagnosed as having diabetes, kidney failure, hepatitis, AIDS (HIV positive) or toxic shock syndrome; or were currently taking (within the last 30 days) immunosuppressive drugs, chemotherapy, systemic antimicrobial or antifungal drugs, or antimicrobials to treat a vaginal infection. Subjects completed a demographic questionnaire and classified themselves into one of the four distinct racial groups: white, black, Hispanic or Asian. The study protocol and informed consent document were reviewed and approved by Hill Top's Institutional Review Board. Informed consent from all subjects was documented before participation in the study.
A sample from each woman was taken near the mid-vagina using a sterile swab. All swabs were taken between days 1 and 10 of the menstrual cycle. A saline-lubricated speculum was used to minimize contamination of the sample by the flora of the labia during entry and withdrawal of the swab. The swab was placed in a sterile cryovial and stored at −70°C until analysis. Upon collection of the vaginal sample, the attending health care practitioner noted any signs of possible genital infections (e.g., discharge, cervicitis or foul odor); none of the subjects in this study had signs of vaginal infection.
From the 3000+ swabs available, we drew random subsets so there would be 15 individuals from each of the five locations (Manitoba, Canada and Ohio, New Jersey, Arizona in the US), and an equal numbers of participants from each of three age groups: 13–18, 19–35 and 36–40 years old (Table 1). It was possible to enroll a full complement of self-declared Caucasian women (n=75). This was also true for black women, except for subjects from Manitoba where too few 13–18 and 35–40 years old self-declared black women enrolled in the previous study (Parsonnet et al., 2005). Consequently, samples from only 69 black women were included in the present study.
Terminal restriction fragment length polymorphism analysis of 16S rRNA genes
For the analysis of terminal restriction fragment length polymorphisms (T-RFLPs) of 16S rRNA genes, internal regions of 16S rRNA genes in each sample were amplified in two separate reactions using fluorescently labeled primer pairs, 8fm–926r and 49f–926r (based on Escherichia coli position). Primer 8fm was labeled with VIC, 49f was labeled with NED and 926r was labeled with 6-FAM (Applied Biosystems, Foster City, CA, USA). DNA amplification was performed as previously described (Zhou et al., 2004) except an annealing temperature of 60°C was used for primer pair 49f–926r. The profiles of terminal restriction fragments from the microbial communities in each sample were determined as follows. A mixture of the two fluorescently labeled amplicons was equally divided and separately digested with MspI and HaeIII, then the digested products were recombined. MspI and HaeIII were chosen because they have been shown empirically and through in silico analyses to provide the greatest resolution of populations likely to be found in vaginal samples. The resulting mixture had six fluorescently labeled terminal restriction fragments from the use of three fluorophores and two restriction enzymes and this allowed for the high resolution of microbial communities. T-RFLP profiles were determined using an ABI PRISM 3100 DNA Analyzer and GeneScan software (Applied Biosystems) as previously described (Coolen et al., 2005) using CST ROX 25–1000 (BioVentures Inc., Murfreesboro, TN, USA) as an internal standard.
Cluster analysis of T-RFLP data
The algorithms described by Abdo et al. (2006) were used for identifying the threshold for defining peaks and for the cluster analysis of T-RFLP data. First, true peaks were identified once a threshold (baseline) had been defined. Second, hierarchical clustering was carried out to identify those fragments with lengths close enough to justly group them in the same length category. Third, the Euclidean distances between T-RFLP profiles were calculated and these were hierarchically clustered based on average linkage (unweighted pair group method and arithmetic mean) and a dendrogram was constructed. Finally, three cluster criteria were employed to identify a statistically meaningful number of groups in the data.
A ‘coverage sampling approach’ (Abdo et al., 2006) was employed to identify the fewest samples that accounted for 85% of the phylotype diversity found within a cluster. In doing so, we assume that in all profiles terminal restriction fragments with a unique size represented a single phylotype. This reduced the total number of samples that needed to be analyzed while at the same time assuring that each cluster was adequately sampled (Abdo et al., 2006). The samples specified were used to construct clone libraries of amplified 16S rRNA genes for subsequent sequence analysis.
Phylogenetic analysis of 16S rRNA gene sequences
Primers FD1f and RD1r (Weisburg et al., 1991) without fluorescent labels were used to generate nearly full-length 16S rRNA amplicons of 16S rRNA genes in vaginal samples. These mixtures of amplicons were used for library construction as described previously (Zhou et al., 2004). We were unable to construct a clone library from one singleton (B1-10) because the yield of genomic DNA was too low.
Approximately 100 clones of each sample were randomly chosen from each library and the cloned DNA fragments were partially sequenced using an ABI 3730 PRISM DNA Analyzer. High-quality sequences with less than 3% uncalled bases and more than 500 bp long were analyzed by using BLAST (Altschul et al., 1997) to identify similar sequences from among the Eubacterial type strains found in the Ribosomal Database Project (RDP; Cole et al., 2003) that were at least 1200 nt long. The most closely related RDP sequences and each sequence from a library were clustered based on genetic similarity using a neighbor joining method (Saitou and Nei, 1987). The resulting clusters were used to define operational taxonomic units (OTUs) in which phylogenetically related clones with ⩾90% sequence similarity to a reference strain were presumed to be members of the same genus, and those whose sequences were ⩾97% similar were considered to be members of the same species. Clones with ⩽90% sequence similarity to any sequence in the RDP were classified as novel organisms. Clones representing each OTU were chosen for detailed phylogenetic analysis by bidirectional sequencing of the entire 16S rRNA gene fragment. These sequences were edited and assembled, then phylogenetic trees were constructed as previously described (Zhou et al., 2004) to depict the phylogenetic relationships that exist among members of the communities. The gene sequences reported in this paper have been deposited in GenBank and were assigned accession numbers AY995236–AY995274.
Statistical modeling and analysis
To evaluate differences in the distribution of microbial community supergroups among Caucasian and black women, we used a method proposed by Schütte et al. (in preparation). We constructed a contingency table (Table 2) with columns of individuals with similar vaginal microbial communities and rows of ethnicity. The counts within each row, associated with each ethnicity, were assumed to be independent and multinomial-distributed. This probabilistic model was then compared to a reduced multinomial model that assumed no ethnic-based differences in the distribution of community supergroups. Comparison between the full model that accounts for ethnicity to the reduced model was carried out using a likelihood ratio test and the bootstrap (Efron and Tibshirani, 1997; Schütte et al., in preparation). Based on the above modeling assumptions, the likelihood function under the full model that accounts for ethnicity was as follows:

where x was a matrix {xij} that represented the counts of individuals having a given microbial community supergroup j associated with the ith ethnicity (i.e., that occupied cell ij of the matrix); N={nW, nB} that represented the total row counts of white and black women, respectively; and Pf was a matrix of parameters, {pij} representing the probabilities of having a microbial community supergroup j associated with the ith ethnicity. As stated above this model assumes that ethnicity impacted the vaginal community structure. There were 16 parameters associated with this model (one parameter per each cell). Under the multinomial model, we estimated only seven parameters per row as the last parameter could be deduced from the others. The maximum likelihood estimates of these parameters were merely the proportions of the cell counts xij to the total sample size of the corresponding row ni, (xij/ni).
For the reduced model, we assumed that the proportion of individuals belonging to a supergroup was the same for both ethnicities. Accordingly, the likelihood of the reduced model could be written as follows:

where x and N were as described above, and Pr was a vector of parameters {pj}, regardless of ethnicity. There were eight parameters associated with this model (one per column), only seven of which need to be estimated, and the maximum likelihood estimate for each of these parameters was

The likelihood ratio test statistic utilized in comparing the two models presented in equation (1) and equation (2) was:

where x and N were as described above, and  and
and  were a matrix of the maximum likelihood estimates of the parameters of the full model and a vector of the maximum likelihood estimates of the parameters of the reduced model, respectively. This likelihood ratio test statistic evaluated the evidence supporting rejection of the reduced model (Equation 2) in favor of the full model (Equation 1), highlighting the importance of ethnicity (if any) in explaining the variation in the count data. To establish the strength of evidence based on this test statistic, we calculated a P-value using the bootstrap (Efron and Tibshirani, 1997).
were a matrix of the maximum likelihood estimates of the parameters of the full model and a vector of the maximum likelihood estimates of the parameters of the reduced model, respectively. This likelihood ratio test statistic evaluated the evidence supporting rejection of the reduced model (Equation 2) in favor of the full model (Equation 1), highlighting the importance of ethnicity (if any) in explaining the variation in the count data. To establish the strength of evidence based on this test statistic, we calculated a P-value using the bootstrap (Efron and Tibshirani, 1997).
Should the reduced model be rejected in favor of the full model we could conclude that there was an effect of ethnicity, and we could proceed to identify those groups that differed most and hence contributed most to the rejection of the reduced model. To accomplish this, we used a standard proportion, pair-wise comparison procedure (Moore and McCabe, 1998).
Results
Classification of vaginal microbial communities
Differences between vaginal microbial communities in Caucasian and black women from North America were identified by analyzing profiles of T-RFLPs of 16S rRNA genes. The data were subjected to cluster analysis to identify similar communities, and the number of clusters (kinds of communities) was independently assessed using three different statistical algorithms as described by Abdo et al. (2006). Among the 144 women sampled, there were 12 kinds of bacterial communities with two or more women, and eight that were represented by a single individual (‘singletons’; Figure 1). The community types that included two or more women account for all those that occur in the populations sampled at a frequency of 0.05 (P=0.99). Thus, we have accounted for those vaginal communities that are commonly found in Caucasian and black women.

Clustering of vaginal microbial communities in Caucasian and black women based on data from T-RFLP analysis of 16S rRNA genes. Groups (clusters) with ⩾2 women designated with a ‘G’ followed by a number. Clusters consisting of a single sample (singletons) are designated with an ‘S’ followed by a number. The samples from each group that were used to construct 16S rRNA gene clone libraries are indicated by asterisks.
To identify the numerically abundant bacterial populations in each kind of community, 16S rRNA gene libraries were constructed from samples that represented 85% of the diversity in each cluster, and from all ‘singletons’. The composition of each sample was determined by phylogenetic analysis of partial 16S rRNA gene sequences (Table 2). In total, 57 clone libraries were analyzed, and approximately 6000 clones (∼90 from each library) were sequenced. Assuming the bacterial numbers in the original samples were on the order of 108 cells per ml of vaginal secretion, then comparatively rare populations present at less than ∼106/ml would not have been sampled and are not reflected in data from the analysis of clone libraries. From these data, it was apparent that the composition of communities of singletons S4 and S7 closely resembled those in group 1 so these were combined to form supergroup I. Likewise, the composition of communities in other groups that were similar to one another were combined to make supergroups II and III (Table 2). The composition of the community in sample W27 differed from others in group 4 by having a high proportion of Veillonella and was therefore removed from the group.
After condensing the community types into supergroups, we found there were eight supergroups with two or more women, and these accounted for 94% of the women sampled.
Composition of vaginal communities
Most vaginal communities (five of eight supergroups) were dominated by organisms that were phylogenetically related to L. iners, L. crispatus, L. jensenii and L. gasseri (Table 2). These accounted for 80% of the women sampled. Overall, L. iners was the most common species of Lactobacillus in women of both races; it was recovered in 66% of the women sampled (95/144), and was the most abundant species in communities of supergroup I, which included 36% (52/144) of the women sampled. Combinations of two or three species of lactobacilli, whose abundances were roughly equal, dominated vaginal communities in supergroups IV, V and VI, and these accounted for 15% of the women sampled. In contrast, communities of supergroup III had low numbers of lactobacilli, and exhibited greater species evenness including high numbers of clones most closely related to Atopobium and genera of the order Clostridiales including Megasphaera, Dialister, Anaerococcus, Finegoldia, Peptostreptococcus and Eubacterium (Figure 2). In addition, 20–30% of the clones from these communities were from miscellaneous novel clades in the phylum Firmicutes. Communities in supergroup VII were dominated by populations most closely related to Streptococcus sp., while those of supergroup VIII had high numbers of clones from a single clade of the family Lachnospiraceae that were unrelated to any named bacterium, but closely related (>97% 16S rRNA gene sequence similarity) to an uncultured bacterium (Figure 2).

A phylogenetic tree showing the relationship of selected phylotypes from vaginal communities of Caucasian and black women to type strains from the order Clostridiales. The phylogenetic tree was constructed using a neighbor-joining algorithm, with Mycoplasma serving as an out-group. Phylotypes in bold font were from this study while phylotypes in italics were from GenBank. Bootstrap values (from 500 replicates) greater than 50% are shown at the branch points, and the bar indicates 10% sequence divergence.
Differences between racial groups
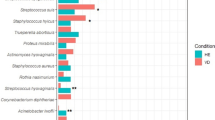
The kinds of microbial communities found in the vaginas of black women differed significantly from those of Caucasian women (Table 2; Figure 3). The incidence of vaginal communities dominated by various species of Lactobacillus (supergroups I, II, IV, V and VI) was less in black women (68%) than in Caucasian women (91%). Conversely, the incidence of communities with a high proportion of phylotypes related to strictly anaerobic bacteria was considerably higher in black women (32%, supergroups III, VII and VIII) than in Caucasian women (8%, supergroups III and VII). Particularly striking was the fact that supergroup III, which includes communities dominated by Atopobium sp. and other strict anaerobes, was four times more common in black women than in Caucasian women. Second, communities dominated by roughly equal numbers of more than one species of Lactobacillus (supergroups IV, V and VI) were found in 25% of Caucasian women but only 6% of black women.

The rank abundances of vaginal community supergroups found in Caucasian and black women. The proportions of Caucasian women are shown as gray bars, and the proportions of black women are shown as black bars.
Visual inspection of the rank-abundance distributions (Figure 3) showed that three of the eight supergroups (II, IV and VI) were more common among Caucasian women, while two supergroups (III and VIII) were more common among black women (Figure 3). To determine whether these differences were statistically significant, we used a method proposed by Schütte et al. (in preparation). The data were summarized in a contingency table wherein individuals were classified based on two factors: community structure and ethnicity (Table 3). Our contention was that if the relative abundances of the microbial communities differed between the two ethnic groups there would be at least one supergroup that differed between them. To test this for all the microbial community supergroups, we assumed that the counts of individuals belonging to each supergroup, within each ethnicity, follow a multinomial distribution that was independent from that of the other ethnicity. Comparison of the full multinomial model that accounted for the effect of ethnicity (Equation 1) to the reduced model that did not (Equation 2) on the basis of likelihood ratios resulted in rejecting the reduced model in favor of the full model. The resulting bootstrap P-value was zero (corresponding to a log-likelihood ratio test statistic equal to 29.26). Accordingly, we concluded that there was a highly significant effect of ethnicity on the frequency of community types in women of the two ethnic groups. Pair-wise comparisons were conducted to identify which of the supergroups accounted for most of the difference attributed to ethnicity (Table 4). The results showed that supergroup III contributed most to the difference between the two ethnicities (P=0.0001), while supergroup IV was also important (P=0.0051). Supergroups II, VI and VIII with P-values of 0.0162, 0.0051 and 0.0749, respectively, had moderate importance in explaining the observed differences in the frequency of community types in women of the two ethnic groups. It is worth noting that the proportion of women in supergroups I, V and VII showed no significant differences between the two ethnic groups.
Diversity of numerically abundant Lactobacillus
The phylogenetic relationships of the Lactobacillus strains were determined by comparing the 16S rRNA gene sequences from this study to those of reference strains sequenced previously. Most lactobacilli found in the vaginal communities were phylogenetically related to L. iners, L. crispatus, L. jensenii and L. gasseri (Figure 4), and were likely to be homofermentative. In contrast, L. vaginalis and L. colehominis were phylogenetically distinct and related to heterofermentative species (Pavlova et al., 2002). The sequence heterogeneity among clones of L. crispatus and L. jensenii was greater than that of L. iners and L. gasseri, suggesting there are evolutionarily divergent subpopulations of L. crispatus and L. jensenii in vaginal communities. Most phylotypes of L. crispatus (∼90%) were most closely related to L. crispatus DSM 20584T (GenBank sequence LCR17362), which is a type strain from Deutsche Sammlung von Mikroorganismen und Zellkulturen. The remainder were most closely related to L. crispatus NCTC4 (GenBank sequence AJ242969) from the National Collection of Type Cultures (London, UK). Thus, there appears to be two distinct lineages that have been classified as L. crispatus, each of which is found in the human vagina. In contrast to L. crispatus and L. jensenii, the clones of L. iners were highly related to one another and to a single reference strain, Lactobacillus sp. LSPY 16329 (data not shown). The occurrence of a virtually clonal lineage of L. iners in different women suggests there might be strong selection for specific phenotypic characteristics that are found in few strains.

A phylogenetic tree based on 16S rRNA gene sequences that shows the relationship of phylotypes recovered in samples of vaginal microbial communities to organisms closely related to Lactobacillus species. Clostridium botulinum E served as an out-group. The phylogenetic tree was constructed using a neighbor-joining algorithm, with Mycoplasma serving as an out-group. Phylotypes in bold font were from this study while phylotypes in italics were from GenBank. Bootstrap values (from 500 replicates) greater than 50% are shown at the branch points, and the bar indicates 10% sequence divergence.
Discussion
In this study, we found that discrete differences exist in the composition of vaginal microbial communities of Caucasian and black women in North America. In total, there were eight distinct types of communities (supergroups) that differed in terms of the kinds and relative abundances of bacterial species, and these accounted for all those that occur at a frequency of 0.05 (P>0.99) in the general populace. Importantly, there were also highly significant differences in the rank abundance of these communities among women in the two racial groups. We hypothesize that these differences in the structure and composition of microbial communities may underlie well-known differences in the susceptibility of women in these racial groups to BV and various vaginal infections (Rajamanoharan et al., 1999; Royce et al., 1999; Whitmore et al., 2005). We specifically postulate that these differences in vaginal communities are accompanied by differences in community resilience, as defined by its ability to resist or absorb perturbations and return to a stable equilibrium state (Peterson et al., 1998). It is largely determined by the ability of indigenous species to adapt to stresses and disturbances, as well as the existence and nature of ecological interactions among species. This notion is consistent with ecological theory and empirical data that indicate not all communities are equally resilient. Thus, if the resilience of a vaginal community is low then transitory changes in the structure of these communities may occur more readily in response to disturbances of various kinds, including menses, sexual intercourse, douching and contraceptive practices. These changes in community structure may include shifts in population densities or species loss, and concomitant changes in community function may occur. These disturbed communities may be more susceptible to invasion by species that are not indigenous to the human vagina including transient species of fecal origin and opportunistic pathogens including Candida. Moreover, we speculate that the disturbed state may itself constitute the clinical syndrome of BV.
There is no information available on the relative resilience of different vaginal microbial communities. However, since the prevalence of BV in black women is higher than in Caucasian women, it would make sense to focus attention on those community types that more commonly occur in black women, such as supergroups III and VIII because they may have lower resilience. The dominant members of these supergroups were Atopobium and various genera in the order Clostridiales. A second hypothesis is that vaginal communities more commonly found in Caucasian women may be more resilient, and therefore more resistant to enduring ecological upsets or invasion by nonindigenous organisms. Such communities are exemplified by supergroups II, IV and VI. Of these, supergroups IV and VI were characterized by the presence of roughly equal numbers of more than one species of Lactobacillus, which may confer increased stability owing to functional redundancy.
It is generally accepted that maintenance of a low pH in the vagina is important for precluding pathogenic organisms, and this function is most often attributed to lactic acid produced by species of Lactobacillus, which are commonly found in the vaginas of healthy women. Consistent with this, we found that lactobacilli constituted ⩾10% of vaginal communities in 79% (107/136) of women. However, Atopobium vaginae also metabolizes glucose via homolactic fermentation (Rodriguez Jovita et al., 1999), and phylotypes related to this species were important constituents of communities in supergroup III. Thus, if one includes members of this genus in the tally, the vaginal communities of all Caucasian women, and all but two black women had high numbers of lactic acid bacteria. The vaginal communities of these two black women were dominated by a novel phylotype that is only distantly related to known genera so there is no basis for speculation about its physiology. Nonetheless, it is clear that the ecological function of vaginal bacterial communities – the formation of lactic acid and maintenance of a low pH environment – was highly conserved among women of both racial groups despite the variations seen in community composition. While this has not been rigorously demonstrated for the human vagina before now, the conservation of function by communities that vary in composition is a common theme in microbial ecology that has been observed in numerous other habitats. Moreover, we found it interesting that only four species of Lactobacillus were numerically important in the communities of women in both racial groups: L. iners, L. crispatus, L. gasseri and L. jensenii. Our results confirm those of Pavlova et al. (2002), who also found two lineages of L. crispatus in the human vagina, raising the specter that one of the two reference strains has been misclassified, or that there is sequence heterogeneity among the 16S rRNA genes in strains of L. crispatus. Nonetheless, the limited number of Lactobacillus phylotypes found in human vagina is somewhat surprising, which leads to the possibility that there are host factors that select for specific organisms, that these species have unusual characteristics that allow them to successfully colonize the vagina, or both.
The strong linkage between high numbers of lactic acid bacteria and a normal microbial community is consistent with Walker's ‘drivers and passengers’ hypothesis of community structure (Walker, 1992). This hypothesis posits that ecosystem function is largely determined by individual ‘driver’ species or guilds (functional groups) of such species and that other species in the community are ‘passengers’ that have minor ecological impact. Driver species strongly influence the species composition and structure of the biological communities in which they and passenger species exist. Under this driver–passenger model, lactic acid bacteria would be considered drivers because they strongly influence the ecosystem by maintaining a low pH through lactic acid production, whereas the other species present would be less influential and constitute passengers with little influence on the ecology of the system. The consideration of such models may prove useful to studies of vaginal microbiology since they provide a framework for understanding the ecology of the vagina, and the possible causes and prevention of ecological upsets such as BV.
Other recent studies also documented the occurrence of Atopobium in vaginal communities of reproductive age and postmenopausal women. For example, Ferris and Masztal (2004) reported that A. vaginae was present in a significant proportion (55%) of women diagnosed as having BV, but in only two of 24 women that had normal vaginal communities. Likewise, Burton et al. (2004) screened 35 postmenopausal women for the occurrence of BV and found A. vaginae in 44% of women with (asymptomatic) BV, while the organism was absent from subjects deemed healthy. Verhelst et al. (2004) and Fredricks et al. (2005) discovered that A. vaginae was frequently detected in subjects with BV. Based on these findings, investigators have implicated A. vaginae in BV. Our results, however, suggest that Atopobium is a commonly encountered constituent of vaginal communities in healthy women. In all of the studies cited above (except ours), the diagnosis of BV was based on the Nugent criteria in which a vaginal smear is Gram-stained and examined to determine the relative abundance of Lactobacillus morphotypes (large Gram-positive rods), and examined for the occurrence and number of lactobacilli that adhere to vaginal epithelial cells. Low numbers or the absence of lactobacilli is the criterion used for the diagnosis of BV. Although Atopobium is a lactic acid bacterium, its cellular morphology is distinctly different from that of Lactobacillus (Rodriguez Jovita et al., 1999). Thus, it seems entirely plausible to us that the women whose communities are dominated by Atopobium and lack appreciable numbers of lactobacilli may be misdiagnosed as having BV. This may partly explain the high incidence of so-called asymptomatic BV that has been reported by many investigators (Schwebke, 2000; Yen et al., 2003). Indeed, given the occurrence in the vagina of lactic acid bacteria that are not species of Lactobacillus, it could be that some patients with reoccurring BV may simply be reflecting their normal vaginal bacterial flora, which are mischaracterized as abnormal. This postulate should be rigorously tested in future studies.
The data obtained in this study showed that a fair proportion of healthy Caucasian and black women also host several fastidious, strictly anaerobic microorganisms that belong to the order Clostridiales. These include appreciable numbers of Lachnospiraceae, Megasphaera, Dialister, Peptoniphilus, Peptostreptococcus and Anaerococcus, as well as many novel bacteria. These populations probably may not have been recovered from samples by using the cultivation methods commonly employed in previous studies of vaginal flora, or routinely used in clinical microbiology laboratories. Given that they appear to constitute more than 5% of bacterial communities in 8% of Caucasian and 32% of black women, efforts are needed to isolate and characterize these organisms to obtain clues to their function in vaginal communities. Importantly, the common occurrence of species classified in the Clostridiales may also have important implications for the clinical diagnosis of BV. Members of this order are notorious for the production of malodorous compounds that include organic acids, amines and thiols through the metabolism of glucose, other carbohydrates and amino acids (Madigan et al., 1997). Complaints of vaginal odor in the absence of infection are not uncommon. For example, Landers et al. (2004) studied 598 women with genital complaints and found that 23% women reported vaginal odor even though there were no outward signs of disease. Till now, the occurrence of odor has been considered abnormal. However, the high number of Clostridiales in many vaginal communities suggests that some degree of odor may be normal for some women, although it could lead to false positives in the diagnosis of BV. Indeed, the presence of amines in vaginal secretions is one of the four criteria used for the diagnosis of BV based on the recommendations on Amsel et al. (1983) that are commonly used in clinical settings. This also suggests that new diagnostic tests for BV that are based on amine production and odor formation should be used with caution (Sonnex, 1995; Hay et al., 2003).
References
Abdo Z, Schütte U, Bent S, Williams C, Forney LJ, Joyce P . (2006). Statistical methods for characterizing diversity in microbial communities by analysis of terminal restriction fragment length polymorphism of 16S rRNA. Environ Microbiol 8: 929–938.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Amsel R, Totten PA, Splegel CA, Chen KCS, Eschenbach D, Holmes KK . (1983). Nonspecific vaginitis: diagnostic criteria and microbial and epidemiologic associations. Am J Med 74: 14–22.
Antonio MA, Hawes SE, Hillier SL . (1999). The identification of vaginal Lactobacillus species and the demographic and microbiologic characteristics of women colonized by these species. J Infect Dis 180: 1950–1956.
Boskey ER, Telsch KM, Whaley KJ, Moench TR, Cone RA . (1999). Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect Immun 67: 5170–5175.
Bradshaw CS, Morton AN, Hodcking J, Garland SM, Morris MB, Moss LM et al. (2006). High recurrence rates of bacteria vaginosis over the course of 12 months after oral metronidazole therapy and factors associated with recurrence. J Infect Dis 193: 1478–1486.
Burton JP, Devillard E, Cadieux P, Hammond J, Reid G . (2004). Detection of Atopobium vaginae in postmenopausal women by cultivation-independent methods warrants further investigation. J Clin Microbiol 42: 1829–1831.
Cole JR, Chai B, Marsh TL . (2003). The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31: 442–443.
Coolen ML, Post E, Davis CC, Forney LJ . (2005). Characterization of microbial communities found in the human vagina by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Appl Environ Microbiol 71: 8729–8737.
Efron B, Tibshirani ARJ . (1997). An Introduction to the Bootstrap. Chapman and Hall/CRC: Boca Raton, FL, USA.
Ferris MJ, Masztal AH . (2004). Use of species-directed 16S rRNA gene PCR primers for detection of Atopobium vaginae in patients with bacterial vaginosis. J Clin Microbiol 42: 5892–5894.
Fredricks DN, Fiedler TL, Marrazzo JM . (2005). Molecular identification of bacterial vaginosis. N Engl J Med 353: 1899–1911.
Goldenberg RL, Klebanoff MA, Nugent R, Krohn MA, Hillier S, Andrews WW . (1996). Bacterial colonization of the vagina during pregnancy in four ethnic groups. Vaginal infections and prematurity study group. Am J Obstet Gynecol 174: 1618–1621.
Haggerty CL, Hillier SL, Bass DC, Ness RB . (2004). Bacterial vaginosis and anaerobic bacteria are associated with endometritis. Clin Infect Dis 39: 990–995.
Hay P, Tummon AOM, Adebiyi A, Adefowora A . (2003). Evaluation of a novel diagnostic test for bacterial vaginosis: ‘the electronic nose’. Int J STD & AIDS 14: 114–118.
Hillier SL, Krohn MA, Cassen E, Easterling TR, Rabe LK, Eschenbach DA . (1995). The role of bacterial vaginosis and vaginal bacteria in amniotic fluid infection in women in preterm labor with intact fetal membranes. Clin Infect Dis 20 (Suppl 2): S276–S278.
Koumans EH, Kendrick JS . (2001). Preventing adverse sequelae of bacterial vaginosis: a public health program and research agenda. Sex Transm Dis 28: 292–297.
Landers DV, Wiesenfeld HC, Heine RP, Krohn MA, Hillier SL . (2004). Predictive value of the clinical diagnosis of lower genital tract infection in women. Am J Obstet Gynecol 190: 1004–1010.
Leitich H, Bodner-Adler B, Brunbauer M, Kaider A, Egarter C, Husslein P . (2003). Bacterial vaginosis as a risk factor for preterm delivery: a meta-analysis. Am J Obstet Gynecol 189: 139–147.
Madigan MT, Martinko JM, Parker JP . (1997). Brock Biology of Microorganisms. Simon & Schuster: Upper Saddle River, NJ, USA.
Marrazzo JM . (2006). A persistently enigmatic ecological mystery: bacterial vaginosis. J Infect Dis 193: 1475–1477.
Martin HL, Richardson BA, Nyange PM . (1999). Vaginal lactobacilli, microbial flora, and risk of human immunodeficiency virus type 1 and sexually transmitted disease acquisition. J Infect Dis 180: 1863–1868.
Moore DS, McCabe GP . (1998). Introduction to the Practice of Statistics, 3rd edn. W.H. Freeman and Co: New York, NY, USA.
Morris M, Nicoll A, Simms I, Wilson J, Catchpole M . (2001). Bacterial vaginosis: a public health review. BJOG 108: 439–450.
Myer L, Denny L, Telerant R, Souza MA, Wright Jr TC, Kuhn L . (2005). Bacterial vaginosis and susceptibility to HIV infection in South Africa women: a nested case-control study. J Infect Dis 192: 1372–1380.
Newton ER, Piper J, Peairs W . (1997). Bacterial vaginosis and intraamniotic infection. Am J Obstet Gynecol 176: 672–677.
Paavonen J . (1983). Physiology and ecology of the vagina. J Infect Dis 40: 31–35.
Parsonnet J, Hansmann M, Delaney M, Modern PA, Du Bois AM, Wieland-Alter W et al. (2005). Prevalence of toxic shock syndrome toxin 1 producing Staphylococcus aureus and the presence of antibodies to this superantigen in menstruating women. J Clin Microbiol 43: 4628–4634.
Pavlova SI, Kilic AO, Kilic SS, So J-S, Nader-Macias ME, Simoes JA et al. (2002). Genetic diversity of vaginal lactobacilli from women in different countries based on 16S rRNA gene sequences. J Appl Microbiol 92: 451–459.
Peterson G, Allen CR, Holling CS . (1998). Ecological resilience, biodiversity, and scale. Ecosystems 1: 6–18.
Rajamanoharan S, Low N, Jones SB, Pozniak AL . (1999). Bacterial vaginosis, ethnicity, and the use of genital cleaning agents: a case control study. Sex Transm Dis 26: 404–409.
Redondo-Lopez V, Cook RL, Sobel JD . (1990). Emerging role of lactobacilli in the control and maintenance of the vaginal bacterial microflora. Rev Infect Dis 12: 856–872.
Rodriguez Jovita M, Collins MD, Sjoden B, Falsen E . (1999). Characterization of a novel Atopobium isolate from the human vagina: description of Atopobium vaginae sp. nov. Int J Syst Bacteriol 49: 573–1576.
Royce RA, Jackson TP, Thorp Jr JM, Hillier SL, Rabe LK, Pastore LM et al. (1999). Race/ethnicity, vaginal flora patterns, and pH during pregnancy. Sex Transm Dis 26: 96–102.
Saitou N, Nei M . (1987). The neighbor-joining methods: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406–425.
Schwebke JR . (2000). Asymptomatic bacterial vaginosis: response to therapy. Am J Obstet Gynecol 183: 1434–1439.
Sewankambo N, Gray RH, Wawer MJ . (1997). HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet 350: 546–550.
Sobel J . (2005). What's new in bacteria vaginosis and trichomoniasis? Infect Dis Clin North Am 19: 387–406.
Sobel JD . (2000). Bacterial vaginosis. Annu Rev Med 51: 349–356.
Sonnex C . (1995). The amine test: a simple, rapid, inexpensive method for diagnosing bacterial vaginosis. Br J Obstet Gynaecol 102: 160–161.
Sweet RL . (1995). Role of bacterial vaginosis in pelvic inflammatory disease. Clin Infect Dis 20: s271–s275.
Taha TE, Hoover DR, Dallabetta GA . (1998). Bacterial vaginosis and disturbances of vaginal flora: association with increased acquisition of HIV. AIDS 12: 1699–1706.
Taha TE, Gray RH, Kumwenda NI, Hoover DR, Mtimavalye LA, Liomba GN et al. (1999). HIV infection and disturbances of vaginal flora during pregnancy. J Acquir Immune Defic Syndr Hum Retrovirol 20: 52–59.
Thorsen P, Jensen IP, Jeune B, Ebbesen N, Arpi M, Bremmelgaard A et al. (1998). Few microorganisms associated with bacterial vaginosis may constitute the pathologic core: a population-based microbiologic study among 3596 pregnant women. Am J Obstet Gynecol 178: 580–587.
Verhelst R, Verstraelen H, Claeys G, Verscharaegen G, Delanghe J, Simaey LV et al. (2004). Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol 4: 1–11.
Walker BH . (1992). Biodiversity and ecological redundancy. Conserv Biol 6: 18–23.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ . (1991). 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697–703.
Whitmore SK, Satcher A, Hu S . (2005). Epidemiology of HIV/AIDS among non-Hispanic black women in the United States. J Natl Med Assoc 97: 19S–24S.
Wiesenfeld HC, Hillier SL, Krohn MA, Amortegui AJ, Heine RP, Landers DV et al. (2002). Lower genital tract infection and endometritis: insight into subclinal pelvic inflammatory disease. Obstet Gynecol 100: 456–463.
Yen S, Shafer MA, Moncada J, Campbell CJ, Flinn SD, Boyer CB . (2003). Bacterial vaginosis in sexually experienced and non-sexually experienced young women entering the military. Obstet Gynecol 102: 927–933.
Zhou X, Bent SJ, Schneider MG, Davis CC, Islam MR, Forney LJ . (2004). Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 150: 2565–2573.
Acknowledgements
We thank Lin Yu, Audra Johnson, Maria G Schneider and Jacob D Pierson for their technical assistance; Stephen J Bent and Ursel Schütte for their valuable advice on analyses of T-RFLP and sequence data; and William Ledger (Weill College of Medicine, Cornell University) for constructive suggestions regarding the manuscript. We gratefully acknowledge the Hilltop Research Facility (Cincinnati, OH) for conducting the clinical study.
Funding/Support: The project described was supported by grants P20RR16448 and P20 RR016454 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. Financial support from the Procter & Gamble Company, Cincinnati, OH was used to design and conduct the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, X., Brown, C., Abdo, Z. et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J 1, 121–133 (2007). https://doi.org/10.1038/ismej.2007.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2007.12
Keywords
This article is cited by
-
Abrupt perturbation and delayed recovery of the vaginal ecosystem following childbirth
Nature Communications (2023)
-
The association between the pre-pregnancy vaginal microbiome and time-to-pregnancy: a Chinese pregnancy-planning cohort study
BMC Medicine (2022)
-
Assessment of bacterial diversity associated with assisted reproductive technologies through next-generation sequencing
Middle East Fertility Society Journal (2022)
-
Evaluation of inhibitory and probiotic properties of lactic acid bacteria isolated from vaginal microflora
Folia Microbiologica (2022)
-
Vaginal Microbiota Is Stable and Mainly Dominated by Lactobacillus at Third Trimester of Pregnancy and Active Childbirth: A Longitudinal Study of Ten Mexican Women
Current Microbiology (2022)
