
Abstract
This study assessed the antihypertensive efficacy of a triple combination, fixed-dose therapy of losartan 50 mg (L50)/hydrochlorothiazide 12.5 mg (H12.5)/amlodipine 5 mg (A5) versus co-administration of L50 plus A5 (L50+A5) in Japanese subjects with uncontrolled essential hypertension. Initially, all subjects received single-blind treatment with L50+A5 for 8 weeks. Subjects whose blood pressure (BP) remained stable within pre-specified limits during the last 4 weeks of L50+A5 administration were randomized (n=327) to double-blind treatment with L50/H12.5/A5 or L50+A5 for 8 weeks. Primary and secondary efficacy endpoints were mean change from baseline to Week 8 in trough diastolic BP (DBP) and trough systolic BP (SBP), respectively. Safety was assessed throughout the study. The treatment difference for L50/H12.5/A5 versus L50+A5 in mean change from baseline in DBP at Week 8 was −1.1 mm Hg (95% confidence interval (CI) −2.7, 0.6; P=0.205). However, the treatment difference in mean change from baseline in SBP at Week 8 was −3.2 mm Hg (95% CI: −5.7, −0.8; P=0.011). A chance imbalance in the change in DBP before randomization between groups was identified in a post-hoc analysis as a major reason for the smaller-than-expected difference in DBP between groups. The overall safety profile was generally similar between groups. In conclusion, treatment with L50/H12.5/A5 for 8 weeks did not demonstrate a significant difference in DBP reduction, but demonstrated a nominally significant difference in SBP reduction, compared with L50+A5. L50/H12.5/A5 was well tolerated. (ClinicalTrials.gov identifier NCT01302691.)
Similar content being viewed by others

Introduction
Approximately 43 million people in Japan suffer from hypertension.1 The majority require a combination of antihypertensive drugs to achieve adequate blood pressure (BP) control. A cross-sectional study conducted in more than 12400 Japanese subjects with hypertension in 2002 found that only 35% of subjects receiving combination therapy with two or more drugs achieved target BP.2 In addition, in a survey of 661 subjects with hypertension conducted at a single center in Japan, only 60% of subjects achieved BP target, although the average number of antihypertensive drugs prescribed was 2.3.3 These results suggest that drugs with stronger antihypertensive effects are needed.
The Japanese Society of Hypertension Guidelines for the Management of Hypertension advocate the use of higher-dose or combination therapy with two or three antihypertensive drugs (including a diuretic) for patients who do not achieve BP targets on antihypertensive monotherapy.1 However, adherence and persistence with antihypertensive treatment is notoriously difficult, as patients often have no notable symptoms of the disease but require life-long treatment with multiple medications, and may experience adverse reactions to drug therapy.4 In addition, many patients with hypertension often have comorbid conditions, such as diabetes and dyslipidemia, which require concomitant treatment with multiple agents.
Fixed-dose antihypertensive drug combinations offer benefits with respect to BP control and compliance.5, 6 A dual, fixed-dose, combination therapy containing the angiotensin receptor blocker losartan and the thiazide diuretic hydrochlorothiazide has been available in Japan since 2006 (PREMINENT; MSD K.K., Tokyo, Japan).
Synergistic antihypertensive effects and reduced drug-related adverse events (AEs) might be expected with concomitant use of losartan, hydrochlorothiazide and a calcium channel blocker as a result of complementary mechanisms of action.7 In Japan, amlodipine is the most frequently used calcium channel blocker, and concomitant use of losartan and amlodipine is one of the recommended combination therapies in the Japanese Society of Hypertension 2014. This study compared the antihypertensive efficacy of a triple-combination, fixed-dose therapy of losartan 50 mg (L50)/hydrochlorothiazide 12.5 mg (H12.5)/amlodipine 5 mg (A5) versus co-administration of L50+A5 in Japanese subjects with essential hypertension uncontrolled with L50+A5, to evaluate the incremental treatment effect with H12.5.
Methods
This was a double-blind, randomized, controlled Phase III trial in Japanese subjects with essential hypertension (protocol number P357; ClinicalTrials.gov identifier NCT01302691), carried out at 30 sites in Japan between February 2011 and April 2012. The study was conducted in accordance with the principles of Good Clinical Practice and the protocol was approved by the appropriate institutional review boards. All subjects provided written informed consent before entering the study.
Subjects
Japanese male and female subjects aged 20–80 years were considered eligible if they had essential hypertension despite receiving a stable regimen of single or dual antihypertensive therapy (up to the highest dose of a single agent, or submaximal doses of dual therapy) for at least 4 weeks before the study. Subjects were required to have a mean trough sitting diastolic BP (DBP) ⩾90 and <110 mm Hg, and systolic BP (SBP) ⩾140 and <200 mm Hg.
Subjects were excluded from the study if they were taking more than two antihypertensive medications, had suspected secondary hypertension or had any history/current evidence of malignant hypertension or hypertensive encephalopathy. Subjects were also ineligible if they had history of clinically significant or relevant cardiovascular disease, known syncopal disorder, bleeding disorders, angioedema, progressive systemic lupus erythematosus, clinically important malabsorption, gastrointestinal resection, malignancy in the past 5 years, uncontrolled diabetes, gout and/or hyperuricemia, anemia, thrombocytopenia, other blood dyscrasia, laboratory parameters out of pre-specified limits, hypersensitivity to study drug components or related drugs, psychiatric disorders, drug or alcohol abuse/dependence, dialysis, pregnancy or lactation. Female subjects of childbearing potential agreed to remain abstinent or use suitable contraception for the duration of the study.
Study design
The study consisted of a screening period of up to 4 weeks, an 8-week single-blind filter period and an 8-week double-blind treatment period. Subjects were randomized 1:1 to double-blind treatment using a computer-generated allocation schedule generated by the clinical biostatistics department of the study sponsor by the permuted block method with a block size of four. Blinding was maintained by means of concomitant administration of matching placebo. Randomization was stratified by the study center.
Treatments
After an initial screening period of up to 4 weeks, eligible subjects discontinued current antihypertensive therapy (tapering over 1 week if necessary) and entered a single-blind filter period during which all subjects received L50+A5 once daily for 8 weeks. Subjects with ⩾75% compliance during the single-blind period (based on patient report) were eligible for randomization to the 8-week double-blind treatment period if their BP had remained within the pre-specified screening limits and reduced by ⩽10 mm Hg (DBP) or ⩽20 mm Hg (SBP) between weeks 4 and 8 of the single-blind period. Eligible subjects were randomized to receive either a single fixed-dose combination tablet of L50/H12.5/A5 or L50+A5 once daily in the morning for 8 weeks.
Assessments
BP and heart rate were measured at each clinic visit 24±2 h after the last study drug administration, with the patient having rested in a sitting position for at least 10 min. AEs, discontinuations, laboratory parameters, vital signs and physical examination were assessed throughout the study and for up to 2 weeks after the end of the double-blind period. Pre-specified safety events of interest were as follows: hypotension reported as an AE; asymptomatic BP decrease (DBP >15 mm Hg or SBP >30 mm Hg); orthostatic hypotension (change from sitting to standing BP >20 mm Hg SBP or >10 mm Hg DBP with symptoms); dizziness; syncope; edema, worsening renal function (increase in serum creatinine >0.5 mg dl−1 from baseline); serum potassium >5.5 mEq l−1 and an increase in serum potassium >0.5 mEq l−1 from baseline; serum potassium <3.5 mEq l−1 and a decrease in serum potassium of >0.5 mEq l−1 from baseline; serum sodium <125 mEq l−1; consecutive elevations in aspartate aminotransferase or alanine aminotransferase more than three times the upper limit of normal; serum uric acid >8.4 mg dl−1 and elevation by 20% from baseline. Compliance was assessed through patient reports (daily diary of time and number of tablets administered) and confirmed by tablet count at each visit.
Endpoints
The primary efficacy endpoint of the study was mean change from baseline in trough sitting DBP with L50/H12.5/A5 compared with L50+A5 after 8 weeks. The secondary efficacy endpoint was mean change from baseline in trough sitting SBP with L50/H12.5/A5 compared with L50+A5 after 8 weeks. Exploratory endpoints included the proportion of subjects responding to treatment (subjects with sitting DBP <90 mm Hg or sitting DBP ⩾90 mm Hg with reduction from baseline ⩾10 mm Hg at week 8) after 8 weeks of treatment and change in mean trough sitting DBP and sitting SBP after 4 weeks of treatment.
Statistical analysis
The primary population for the efficacy evaluation was the full analysis set population, defined as all randomized subjects who received at least one dose of the study medication and had at least one post-randomization observation after at least one dose of the study medication. The primary and secondary efficacy endpoints were analyzed using a constrained longitudinal analysis8 model with terms for treatment, time and treatment-by-time interaction. Missing data were not explicitly imputed. The week 4 results were also obtained from the same model. Multiplicity across the primary and secondary efficacy endpoints was adjusted using a sequential (step-down) testing procedure.
For the analysis of the proportion of responders at week 8, five sets of imputations were made with missing data imputed, based on the constrained longitudinal analysis model above. Each of the imputed data sets was analyzed using a logistic regression with a term for treatment and baseline mean trough sitting DBP as a covariate. Parameter estimates on log odds ratio were combined using the asymptotic theory of Robins and Wang,9 with Kenward and Roger10 degrees of freedom used to construct the confidence interval (CI) and calculate the P-value.
Safety analysis was performed in the all-subjects-as-treated population, which was defined as all randomized subjects who received at least one dose of study treatment. P-values and 95% CIs for between-treatment differences in the percentage of subjects with pre-specified safety events of interest were calculated using the method of Miettinen and Nurminen.11 All other AEs and safety events were assessed via point estimates with 95% CIs for between-group comparisons or point estimates by treatment group. All statistical tests were two-sided with a significance level of 5%.
It was planned that 326 subjects would be randomized equally to treatment with L50/12.5/A5 or L50+A5 to ensure 146 completers per treatment arm. This sample size was designed to detect a between-treatment difference of −2.9 mm Hg (s.d. estimate 7.6) and −4.7 mm Hg (s.d. 12.2) in mean trough sitting DBP and SBP, respectively, with 90% power, all at a two-sided significance level of 0.05. The power calculation was based on the inverse probability-weighted complete cases approach described by Lu et al.,12 assuming a rate of discontinuation of 10%.
All statistical analyses were performed by the Clinical Biostatistics Department of MSD K.K.
Results
Subjects
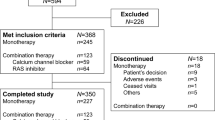
The flow of subjects through the study is shown in Figure 1. In total, 707 subjects were screened. Of these, 327 subjects were randomized to double-blind treatment with L50/H12.5/A5 (n=164) or L50+A5 (n=163). All 327 subjects were included in both the full analysis set and all-subjects-as-treated populations. Baseline characteristics were generally comparable between treatment groups (Table 1). Average compliance was 99.5% in each group and no instance of patient compliance was <75%.

Flow of subjects through the study. A5, amlodipine 5 mg; AE, adverse event; H12.5, hydrochlorothiazide 12.5 mg; L50, losartan 50 mg.
Efficacy
The treatment difference for L50/H12.5/A5 versus L50+A5 in mean change from baseline in trough sitting DBP at week 8 was −1.1 mm Hg (95% CI −2.7, 0.6; P=0.205; Table 2). The reduction in trough sitting DBP was numerically greater with L50/H12.5/A5 than with L50+A5, but not statistically significant. The treatment difference in mean change from baseline in trough sitting SBP was −3.2 mm Hg (95% CI −5.7, −0.8; P=0.011; Table 2). The reduction in trough sitting SBP was greater with L50/H12.5/A5 than with L50+A5 and achieved nominal significance, although statistical significance could not be declared due to the pre-specified strategy for multiplicity adjustment.
The estimated treatment differences between L50/H12.5/A5 and L50+A5 in mean change from baseline in trough sitting DBP and SBP at week 4 were −1.5 mm Hg (95% CI −3.0, −0.0; P=0.049) and −2.7 mm Hg (95% CI −4.9, −0.5; P=0.014), respectively (Supplementary Table 1). The proportion of subjects responding to treatment after 8 weeks was 68.3% (95% CI 60.8, 74.9) in the L50/H12.5/A5 group and 62.1% (95% CI 54.4, 69.2) in the L50+A5 group. The odds ratio of responding to treatment was 1.18 (95% CI 0.73, 1.90; P=0.510).
A substantially larger mean increase in trough sitting DBP was observed during the last 4 weeks of the 8-week filter period before randomization with the L50+A5 group, compared with the L50/H12.5/A5 group (3.5 versus 1.8 mm Hg, respectively; Figure 2 and Supplementary Table 2). Incidentally, subjects who experienced a greater increase in trough sitting DBP before randomization had a greater decrease in trough sitting DBP after randomization to week 8 (Supplementary Figure 1).

Change from week 0 baseline in mean trough sitting DBP. A5 amlodipine 5 mg; DBP, diastolic blood pressure; H12.5, hydrochlorothiazide 12.5 mg; L50, losartan 50 mg.
Two post-hoc efficacy analyses were performed. In the first, a post-hoc analysis using a constrained longitudinal analysis model with additional factors of change in trough sitting DBP during the last 4 weeks of the filter period and its interaction with time resulted in a least squares mean difference (95% CI) in trough sitting DBP of −1.5 (−3.1, 0.1) mm Hg for the entire full analysis set population and −1.6 (−3.3, 0.1) mm Hg for subjects with change in mean trough sitting SBP/DBP within 20/10 mm Hg before randomization (Supplementary Table 3). When baseline DBP was defined as the average of the two sitting DBP measurements before randomization in the primary constrained longitudinal analysis model (the second post-hoc analysis), least squares mean (95% CI) was −1.8 (−3.4, −0.2) mm Hg for the entire full analysis set population and −2.0 (−3.8, −0.3) mm Hg in subjects with change in mean trough sitting SBP/DBP within 20/10 mm Hg before randomization (Supplementary Table 4).
Safety
A similar proportion of subjects experienced AEs in each treatment group (Table 3 and Supplementary Table 5). One subject in each treatment group experienced a serious AE. In the L50/H12.5/A5 treatment group, one subject experienced clavicle fracture, pneumothorax and rib fracture (not considered drug related); in the L50+A5 treatment group, one subject died (sudden cardiac death), also not considered drug related. More subjects receiving L50/H12.5/A5 (11.6%) experienced drug-related AEs than in the L50+A5 group (3.7%); however, the only drug-related AE with an incidence ⩾2% was an increase in serum uric acid (L50/H12.5/A5: 7/164 patients (4.3%), L50+A5: 2/163 patients (1.2%)). Two subjects taking L50/H12.5/A5 discontinued due to AEs, compared with none in the L50+A5 group. Discontinuations were a result of a non-serious AE (hypokalemia, which was considered to be drug related) and a serious AE (the patient with clavicle fracture, pneumothorax and rib fracture, which was not considered to be drug related).
There was no statistically significant difference between the treatment groups in terms of percentage of subjects experiencing any pre-specified safety events of interest (Table 4). However, the percentage of subjects with serum uric acid >8.4 mg dl−1 and elevation by >20% from baseline was numerically greater, although not statistically significantly greater, in the L50/H12.5/A5 group (3.7%) than the L50+A5 group (0.6%), P=0.058. Mean baseline serum uric acid (s.d.) in the L50+A5 (n=163) and L50/H12.5/A5 (n=163) groups was 5.6 (1.3) and 5.5 (1.2) mg dl−1, respectively, and the change from baseline at 8 weeks was −0.01 (0.6) and 0.6 (0.8) mg dl−1, respectively.
Discussion
The present study did not demonstrate a significant difference in DBP reduction with L50/H12.5/A5 for 8 weeks compared with L50+A5, but demonstrated a nominally significant difference in SBP reduction compared with L50+A5. L50/H12.5/A5 was well tolerated with a similar AE profile to L50+A5.
The efficacy findings were somewhat unexpected as other triple-combination, fixed-dose therapies containing hydrochlorothiazide and amlodipine with olmesartan, valsartan, or aliskiren have shown greater BP-lowering activity compared with dual combination therapy with any two of their components in subjects with stage-2 hypertension (albeit when administered at maximum doses of each component).13 A recent study in Japanese subjects with uncontrolled essential hypertension compared L50/H12.5/A5 with a dual fixed-dose combination therapy with L50/H12.5 and showed that mean trough sitting DBP was reduced to a significantly greater extent over 8 weeks in subjects treated with L50/H12.5/A5 (P<0.001).14 The current trial results are not consistent with these prior studies. Therefore, we explored potential reasons for this inconsistency.
In the present study, the treatment difference in the primary efficacy endpoint of change from baseline in mean trough sitting DBP at week 8 was smaller than expected and superiority of L50/H12.5/A5 versus L50+A5 group was not demonstrated. Even though there was a difference of 1 mm Hg between the two groups in mean sitting DBP at baseline (L50/H12.5/A5: 95.3±4.3 mm Hg, L50+A5: 96.3±4.3 mm Hg), the analysis model adjusts for baseline by placing a constraint that the true baseline mean is equal between treatments. Hence, it is unlikely that the smaller-than-expected treatment difference is due to the imbalance in mean DBP at baseline. Instead, it may be due to a large variability in BP during the last 4 weeks of the 8-week filter period before randomization (Figure 2). Compared with the primary analysis, the post-hoc analysis adjusting for the DBP change during the last 4 weeks of the filter period resulted in a slightly larger treatment difference in DBP change (Supplementary Table 3). The post-hoc analysis with baseline defined as the average of the two sitting DBP measurements before randomization also resulted in a greater treatment difference in DBP change compared with the difference from the primary analysis (Supplementary Table 4). These findings suggest that the variability in DBP during the pre-randomization period was not fully controlled by defining the single timepoint of the randomization visit alone as baseline, which could be considered a limitation of the present study.
Data from post-hoc analyses are consistent with observations in previous factorial studies that have demonstrated the additional hydrochlorothiazide efficacy in addition to angiotensin receptor blocker plus calcium channel blocker.14 Therefore, it remains plausible that the addition of H12.5 to L50 and A5 will have the potential to contribute to a reduction in mean trough DBP beyond the values obtained in this study.
The current study also investigated the effect of L50/H12.5/A5 compared with L50+A5 on mean trough sitting SBP. SBP is important as an independent and strong predictor of risk of cardiovascular and renal disease.15 A nominally significant estimated difference between groups in mean change from baseline in sitting SBP at week 8 in favor of L50/H12.5/A5 was demonstrated (Table 2).
The favorable safety profile of dual therapy with amlodipine/losartan or losartan and hydrochlorothiazide has been previously reported in Japanese hypertensive subjects.16, 17, 18, 19 The current study revealed a comparable safety and tolerability profile for L50/H12.5/A5 compared with dual L50+A5. Over the 8-week double-blind phase of the study, the overall safety profile of the two treatment regimens was generally similar. A greater proportion of subjects receiving L50/H12.5/A5 than L50+A5 experienced AEs that were deemed to be drug related (11.6% versus 3.7%); however, none of these AEs were serious and there was no statistically significant difference between the treatment groups in terms of the percentage of subjects experiencing any pre-specified safety events of interest. Although assessment of safety in this study was limited by the short duration (8 weeks), the long-term (1-year) safety of L50/H12.5/A5 has previously been demonstrated in a similar population of Japanese subjects with uncontrolled essential hypertension.14 In general, triple-combination, fixed-dose drugs have been shown to be well tolerated with a low incidence of AEs, the most common being peripheral edema related to amlodipine.13
Historically, diuretic use in Japan has remained low due to concerns about negative effects on metabolic parameters.2, 17 In the present study, the percentage of subjects with elevated serum uric acid levels was greater in the L50/H12.5/A5 group than in the L50+A5 group. The characteristic serum uric acid-excreting effect of losartan might have been expected to offset hyperuricemia typically associated with hydrochlorothiazide as observed in other studies. Indeed, during the PALM-1 Extension Study, a 3-year safety analysis of a dual fixed-dose losartan/hydrochlorothiazide combination in Japanese subjects with uncontrolled hypertension, the losartan/hydrochlorothiazide combination appeared to minimize diuretic-related AEs.18 Despite this, small numbers of subjects did experience AEs, such as hypokalemia or hyperuricemia, and careful monitoring of blood parameters is required. Interestingly, a recent study comparing dual fixed-dose combinations of losartan/hydrochlorothiazide and losartan/amlodipine in Japanese subjects with uncontrolled hypertension found that losartan/hydrochlorothiazide was associated with significantly increased serum uric acid; however, this effect was only evident in those subjects with low baseline serum uric acid levels (<5.6 mg dl−1).20 Notably, subjects in the present study had baseline serum uric acid levels of 5.5–5.6 mg dl−1.
In conclusion, the present study has demonstrated a numerically greater but not statistically significant DBP reduction with L50/H12.5/A5 versus L50+A5, and a nominally significant SBP reduction with L50/H12.5/A5 versus L50+A5 in Japanese subjects. The triple combination therapy was well tolerated.
References
Shimamoto K, Ando K, Fujita T, Hasebe N, Higaki J, Horiuchi M, Imai Y, Imaizumi T, Ishimitsu T, Ito M, Ito S, Itoh H, Iwao H, Kai H, Kario K, Kashihara N, Kawano Y, Kim-Mitsuyama S, Kimura G, Kohara K, Komuro I, Kumagai H, Matsuura H, Miura K, Morishita R, Naruse M, Node K, Ohya Y, Rakugi H, Saito I, Saitoh S, Shimada K, Shimosawa T, Suzuki H, Tamura K, Tanahashi N, Tsuchihashi T, Uchiyama M, Ueda S, Umemura S . The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2014). Hypertens Res 2014; 37: 253–387.
Mori H, Ukai H, Yamamoto H, Saitou S, Hirao K, Yamauchi M, Umemura S . Current status of antihypertensive prescription and associated blood pressure control in Japan. Hypertens Res 2006; 29: 143–151.
Ohta Y, Tsuchihashi T, Kiyohara K . Relationship between blood pressure control status and lifestyle in hypertensive outpatients. Intern Med 2011; 50: 2107–2112.
Erdine S . How do compliance, convenience, and tolerability affect blood pressure goal rates? Am J Cardiovasc Drugs 2012; 12: 295–302.
Panjabi S, Lacey M, Bancroft T, Cao F . Treatment adherence, clinical outcomes, and economics of triple-drug therapy in hypertensive patients. J Am Soc Hypertens 2013; 7: 46–60.
Sakima A, Ohshiro K, Nakada S, Yamazato M, Kohagura K, Nakamoto M, Tana T, Ohya Y . Switching therapy from variable-dose multiple pill to fixed-dose single-pill combinations of angiotensin II receptor blockers and thiazides for hypertension. Clin Exp Hypertens 2011; 33: 309–315.
Neutel JM, Smith DH . Hypertension management: rationale for triple therapy based on mechanisms of action. Cardiovasc Ther 2013; 31: 251–258.
Liang K-L, Zeger SL . Longitudinal data analysis of continuous and discrete responses for pre-post designs. Sankyâ 2000; 62: 134–148.
Robins JM, Wang N . Inference for imputation estimators. Biometrika 2000; 87: 113–124.
Kenward MG, Roger JH . Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 1997; 53: 983–997.
Miettinen O, Nurminen M . Comparative analysis of two rates. Stat Med 1985; 4: 213–226.
Lu K, Mehrotra DV, Liu G . Sample size determination for constrained longitudinal data analysis. Stat Med 2009; 28: 679–699.
Taylor AA, Ragbir S . Three in one: safety, efficacy, and patient acceptability of triple fixed-dose combination medicine in the management of hypertension. Patient Prefer Adherence 2012; 6: 555–563.
Rakugi H, Tsuchihashi T, Shimada K, Numaguchi H, Nishida C, Yamaguci H, Shirakawa M, Azuma K, Fujita KP . Efficacy and safety of triple therapy with losartan 50mg/hydrochlorothiazide 12.5mg/amlodipine 5mg in Japanese patients with essential hypertension: a randomized, controlled trial. Clin Exp Hypertens 2014 (e-pub ahead of print 1 October 2014; doi:10.3109/10641963.2014.954712).
He J, Whelton PK . Elevated systolic blood pressure as a risk factor for cardiovascular and renal disease. J Hypertens Suppl 1999; 17: S7–S13.
Maeda K, Adachi M, Kinoshita A, Koh N, Miura Y, Murohara T . Efficacy and safety of the losartan-hydrochlorothiazide combination tablet in patients with hypertension uncontrolled by angiotensin II receptor antagonist therapy: the Aichi Research on Combination therapy for Hypertension (ARCH) Study. Intern Med 2012; 51: 1167–1175.
Ohta Y, Tsuchihashi T, Onaka U, Eto K, Tominaga M, Ueno M . Long-term compliance with salt restriction in Japanese hypertensive patients. Hypertens Res 2005; 28: 953–957.
Kita T, Yokota N, Ichiki Y, Ayabe T, Etoh T, Tamaki N, Kato J, Eto T, Kitamura K . Three-year safety and effectiveness of fixed-dose losartan/hydrochlorothiazide combination therapy in Japanese patients with hypertension under clinical setting (PALM-1 Extension Study). Clin Exp Hypertens 2012; 34: 498–503.
Oshikawa J, Toya Y, Morita S, Taguri M, Hanaoka K, Hasegawa T, Kaizu K, Kamata K, Kobayashi S, Ohtake T, Sato T, Yasuda G, Kimura K, Umemura S . Angiotensin receptor blocker (ARB)-diuretic versus ARB-calcium channel blocker combination therapy for hypertension uncontrolled by ARB monotherapy. Clin Exp Hypertens 2014; 36: 244–250.
Oshikawa J, Toya Y, Morita S, Taguri M, Hanaoka K, Hasegawa T, Kaizu K, Kamata K, Kobayashi S, Ohtake T, Sato T, Yasuda G, Kimura K, Umemura S . Angiotensin receptor blocker (ARB)-diuretic versus ARB-calcium channel blocker combination therapy for hypertension uncontrolled by ARB monotherapy. Clin Exp Hypertens 2013; 36: 244–250.
Acknowledgements
P357 study investigators: S. Morikawa (Clinic Horikawa), Y. Ohashi (Medical Corporation Kouhoukai Tokyo Ekimae-building Clinic), M W Hwang (Koh Internal Medicine Clinic), S. Inoue (Heishinkai Medical Group Incorporated OCROM Clinic), R. Yuyama (Rakusai Newtown Hospital), H. Oda (Oda Clinic), H. Konishi (Yamamoto Hospital), T. Shiraiwa (Shiraiwa medical clinic), S. Yano (Maebashi Hirosegawa Clinic), T. Hoshina (Hoshina Clinic Medical Corporation Keneikai), O. Matsuoka (Heishinkai Medical Group Incorporated ToCROM Clinic), T. Ogasawara (Medical Corporation Sai Tadayoshi Kai SAIKATSU CLINIC), Y. Noguchi (Noguchi Naika Clinic), K. Mizuyama (Doujin Memorial Foundation Meiwa Hospital), M. Saito (Kaijo Bldg. Clinic), T. Doi (Medical Corporation Sanshukai Doi Internal Medicine Clinic), H. Beppu (Medical Corporation Beppunaika Clinic), Y. Shimizu (Sapporo Higashi Tokushukai Hospital), Y. Tatsuoka (Tatsuoka Neurology Clinic), Y. Hayakawa (Medical Corporation Meiwa Hospital), M. Ozaki (Social Medical Corporation Reimei-Kai Kitade Hospital), M. Irahara (Irahara Clinic), N. Kusunose (Kusunose Surgery Clinic), T. Nakamura (Nakamuratadashi Naikajunkankika Clinic), T. Eguchi (Eguchi Clinic), Y. Nishi (NAGATA HOSPITAL), M. Tsujimoto (The Veritas Hospital), S. Kouno (Asunaro Naika Clinic), A. Yamauchi (Medical Care Law person corporation Rikeikai Suruga Clinic), S. Morizono (Morizono Medical Clinic). Medical writing and editorial assistance was provided by Melanie Jones of Prime Healthcare, Knutsford, Cheshire, UK. This assistance was funded by Merck & Co. Inc., Kenilworth, NJ, USA. We thank Mary E Hanson, PhD, of Merck & Co. Inc., Kenilworth, NJ, USA, for scientific input and assistance with writing and editorial support. Analysis of the study data and opinions, conclusions and interpretation of the data are the responsibility of the authors. We also thank William Malbecq, Kristel Vandormael and Rachid Massaad of MSD Belgium for providing statistical expertise; Emanuela Santoro of Merck & Co. Inc., Kenilworth, NJ, USA, for assistance with study conduct; Yoichi Inoue, MD, and Hiromi Iwata of MSD K.K. for assistance with conduct of the study; Sachiko Yoshimoto, MD, of MSD K.K. for assistance with study design and conduct; and the Primary Investigators for conduct of the study. The study was funded by Merck Sharp & Dohme K.K., Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
HR has received honoraria and/or fees for promotional materials from Astellas Pharma, Daiichi Sankyo, Kyowa Hakko Kirin, Nippon Boehringer Ingelheim, Novartis Pharma, Takeda Pharmaceutical and MSD KK, and has received research funding from Astellas Pharma, Daiichi Sankyo, Kyowa Hakko Kirin, Mitsubishi Tanabe Pharma, MSD KK, Nippon Boehringer Ingelheim, Novartis Pharma, Pfizer Japan and Takeda Pharmaceutical. TT has received honoraria and/or fees for promotional materials from Dainippon-Sumitomo Co. and MSD KK, and has received research funding from MSD KK. KS has received honoraria and/or fees for promotional materials from Daiichi Sankyo, Takeda Pharmaceutical, Dainippon-Sumitomo Co., Novartis Pharma KK and MSD KK. HN, CN, HY, MS and KA are employees of MSD K.K and may own stock or hold stock options in the company. KPF was an employee of Merck & Co., Inc., Kenilworth, NJ, USA, at the time the study was conducted.
Additional information
Supplementary Information accompanies the paper on Hypertension Research website
Supplementary information
Rights and permissions
About this article

Cite this article
Rakugi, H., Tsuchihashi, T., Shimada, K. et al. Add-on effect of hydrochlorothiazide 12.5 mg in Japanese subjects with essential hypertension uncontrolled with losartan 50 mg and amlodipine 5 mg. Hypertens Res 38, 329–335 (2015). https://doi.org/10.1038/hr.2015.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2015.3
Keywords
This article is cited by
-
The benefits of angiotensin-converting enzyme inhibitors/angiotensin II receptor blockers combined with calcium channel blockers on metabolic, renal, and cardiovascular outcomes in hypertensive patients: a meta-analysis
International Urology and Nephrology (2018)
-
Sacubitril/valsartan in the treatment of arterial hypertension: an unaccomplished promise?
Hypertension Research (2017)
-
Antihypertensive medication and risk of kidney stones: a Canadian wake-up call
Hypertension Research (2017)
-
The effects of increasing calcium channel blocker dose vs. adding a diuretic to treatment regimens for patients with uncontrolled hypertension
Hypertension Research (2017)
-
The efficacy and long-term safety of a triple combination of 80 mg telmisartan, 5 mg amlodipine and 12.5 mg hydrochlorothiazide in Japanese patients with essential hypertension: a randomized, double-blind study with open-label extension
Hypertension Research (2017)
