
Abstract
Vascular endothelial cells are exposed to an acidic pH, and CXC chemokine receptor type 4 (CXCR4) is a key protective molecule against acidosis. We investigated the effect of coupling factor 6 (CF6), a novel proton import activator, on CXCR4 signaling and its molecular mechanism. CF6 decreased CXCR4 expression in human umbilical vein endothelial cells (HUVECs) in a time- and dose-dependent manner. Pretreatment with small interfering RNA (siRNA) for hypoxia-inducible factor (HIF)-1α or PP1, a specific c-Src inhibitor, attenuated the CF6-induced decrease in CXCR4 without affecting CF6-induced intracellular acidosis. Chromatin immunoprecipitation revealed that CF6 enhanced the interaction between HIF-1α and the CXCR4 promoter at the hypoxia response element. CF6 also enhanced protein–protein interactions between phospho-c-Src and histone deacetylase 3 (HDAC3), but did not affect the binding of HDAC3 to the CXCR4 promoter at the hypoxia response element. Apoptotic cells, as measured by an Annexin-V-FITC Propidium Iodide Kit, were increased by CF6 in normoxia and hypoxia at 24 h; however, this increase was abolished by pretreatment with either siRNA for HIF-1α or the CXCR4 ligand. The coronary arteries and perivascular tissues obtained from CF6-overexpressing transgenic mice showed a lower expression of CXCR4 in the heart, increased wall thickness and infiltration of CD16-positive, CD206-positive or apoptotic cells. CF6 decreases CXCR4 expression through both HIF-1α- and c-Src-mediated mechanisms in vascular endothelial cells. Because CXCR4 has an important role in survival function, CF6 may have a role in the progression of arteriosclerosis via these complex mechanisms.
Similar content being viewed by others


Introduction
CXC chemokine receptor type 4 (CXCR4) is expressed in vascular endothelial cells, as well as in hematopoietic and epithelial cells; CXCR4 binds the chemokine CXCL12 (stromal cell-derived factor-1, SDF-1), which is constitutively produced by bone marrow stromal cells and epithelial cells in several other organs, including the lymph nodes, liver, lung, spleen, heart, skin, kidney and brain.1 The binding of CXCL12 to CXCR4 activates several divergent intracellular pathways, such as extracellular signal-regulated protein kinase, Src family kinases, phosphatidylinositol 3-kinase and Akt, through Giα-protein- and G-protein-independent Janus kinase/signal transducers, as well as activators of transcription 3 signal and p38 mitogen-activated protein (MAP) kinase.2, 3, 4 Activation of the chemokine receptor CXCR4 regulates chemotaxis, survival, proliferation, gene transcription and intracellular calcium flux in various types of cells.1 Vascular endothelial cells are exposed to an acidic pH under a variety of pathological and physiological conditions, including ischemia and/or hypoxia and shear stress,5 and CXCR4 was recently reported as a key molecule in the acidosis response.6
We recently identified coupling factor 6 (CF6) as a novel activator for a proton importer that elicits a sustained decrease in intracellular pH.7 CF6 activates plasma membrane ATP synthase (F1Fo complex); the molecular rotary motor, F1-ATPase, forcefully hydrolyzes ATP in a mode that is the reverse of the mode used in mitochondria. This hydrolysis inversely rotates the Fo motor against the original clockwise direction, resulting in proton import.7 CF6 suppresses prostacyclin generation via inhibition of cytosolic phospholipase A28 and suppresses nitric oxide (NO) generation via upregulation of asymmetric dimethylarginine, an endogenous competitive inhibitor of NO synthase.9 Prostacyclin and NO are recognized as the major mediators of vascular homeostasis maintenance, and a decrease in these compounds is implicated in endothelial dysfunction. In the present study, we examined the role of CF6 in endothelial dysfunction via the CXCR4 signaling pathway. We report here that CF6 attenuates CXCR4 expression through hypoxia-inducible factor (HIF)-1α- and c-Src-mediated pathways and promotes apoptosis in vascular endothelial cells. In CF6-overexpressing transgenic mice (TG), wall thickness and inflammation are increased in small coronary vessels.
Methods
Materials
The QIA shredder and RNeasy Protect Mini Kit were obtained from Qiagen (Valencia, CA, USA). Human CXCR4, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers, the TaqMan probe, TaqMan reverse transcription reagent, and TaqMan universal polymerase chain reaction (PCR) master mix were obtained from Applied Biosystems (Foster City, CA, USA). HIF-1α annealed siGENOME SMART pool (cat. no. L-004018-00-0005), siCONTROL Non-Targeting siRNA no.1 (cat. no. D-001210-01) and DharmaFECT 1 (cat. no. T-2002-01) were all obtained from Dharmacon (Lafayette, CO, USA). The HuMedia-EG2 Kit was purchased from Kurabo (Osaka, Japan). The AnaeroPack and hypoxic chamber were obtained from Mitsubishi Gas Chemistry (Tokyo, Japan). The Annexin V-FITC Apoptosis Detection Kit was obtained from Abcam (Cambridge, MA, USA). The ApopMark Apoptosis Detection Kit was obtained from Exalpha Biologicals (Maynard, MA, USA). Human CF6 was obtained from Phoenix Pharmaceuticals (Belmont, CA, USA). Human SDF-1 was obtained from R&D Systems (Minneapolis, MN, USA). RIPA lysis buffer and human CXCR4, HIF-1α, CD206, CD16 and GAPDH antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Human histone deacetylase 3 (HDAC3) antibody was obtained from Cell Signaling Technology (Danvers, MA, USA). Polyvinylidene difluoride membrane was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Enhanced chemiluminescence plus detection systems were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA). All other reagents were of the finest grade available from Sigma Chemical (St Louis, MO, USA).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were cultured in HuMedia-EG2 (complete media) at 37 °C under 5% CO2. HUVECs from the second to sixth passages were used for the study. After treatment with various molecules, the viability of the cells, which was determined by trypan blue excretion, was generally >95%.
Experimental animals
All procedures were approved by the ethics committee for animal experimentation of Hirosaki University Graduate School of Medicine (Hirosaki, Japan). The investigation protocols conformed to either the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health or the Directive 2010/63/EU of the European Parliament. TG were generated as reported elsewhere.10 Briefly, the introduced gene product was released outside the cells by a secretion signal upstream from the mature human CF6 (Asn33-Ala108) and was expressed in overall tissues by the human elongation factor 1α promoter. CF6 mRNA expression was upregulated by 1.94±0.27 times in overall tissues, including the heart and kidney, in the TG compared with wild-type (WT) mice, and the plasma level of total CF6 was two times as high in the TG as in the WT.11 The intracellular pH value, as measured by 31P-magnetic resonance spectroscopy, was decreased by 0.1 U in skeletal muscle and liver of the TG.11 For histological analysis, mice were killed by cervical dislocation under anesthesia with intraperitoneal administration of 0.3 mg kg−1 medetomidine, 4 mg kg−1 midazolam and 5 mg kg−1 butorphanol. Neither blood pressure nor heart rate differed between the TG and WT.
Determination of gene expression
Total RNA was extracted from the cells using the QIAamp RNA Kit (Qiagen, Valencia, CA, USA). A two-step reverse transcription-PCR was carried out according to the protocol supplied with the TaqMan Gold RT–PCR Kit (Applied Biosystems, Foster City, CA, USA). The standard curves for CXCR4 and GAPDH were linear between 0.1 and 250 ng μl−1 total RNA. Values were averaged from duplicate data and normalized to human GAPDH.
Determination of protein expression and immunoprecipitation
Tissue and cell samples were homogenized in RIPA lysis buffer (20 mmol l−1 Tris-HCl (pH 7.5), 150 mmol l−1 NaCl, 1 mmol l−1 EDTA, 1 mmol l−1 EGTA, 1% Triton X-100, 1% glycerol, 1 mmol l−1 dithiothreitol and 0.5 mmol l−1 phenylmethylsulfonyl fluoride). Samples were mixed with Laemmli buffer that contained 5% β-mercaptoethanol and were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For immunoprecipitation, samples were incubated with anti-phospho-c-Src antibody and a protein A/G plus-agarose immunoprecipitation reagent overnight at 4 °C. After centrifugation, the immunoprecipitates were applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Protein was electrophoretically transferred to a polyvinylidene difluoride membrane and incubated with the primary antibodies for CXCR4, HDAC3 and GAPDH at 4 °C overnight. The protein bands were detected by enhanced chemiluminescence plus detection systems. Densitometric analysis was performed with Scion image software (Informer Technologies, Inc., Copthall Roseau Valley, Dominica), and the relative ratio to the protein bands was calculated for each sample.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed using HUVECs treated with CF6 at 10−7 M for 7 h. Briefly, 5 × 106 cells were fixed with 1% paraformaldehyde for 5 min and crosslinked; the crosslinked chromatin was then sheared by sonication. The chromatin solution was incubated with 5 μg of anti-HIF-1α and anti-HDAC3 antibodies or control immunoglobulin G and protein A/G plus-agarose immunoprecipitation reagent overnight at 4 °C. Immunoprecipitates were eluted from the protein A/G plus-agarose reagent, and supernatants were treated with RNase A and proteinase K, extracted and precipitated. The pellets were then resuspended in 30 μl of H2O and subjected to 40 cycles of PCR amplification with the following primers specific to the human CXCR4 promoter as follows: sense, 5′-GGGCTTCCCAAGCCGCGCACCT-3′ and antisense, 5′-CTGCCGCAGCCAACAAACTGAAGT-3′.
Histological analysis
Histological analysis was performed by fixing hearts overnight in 10% phosphate-buffered formalin; the samples were processed into paraffin blocks for section. Serial 5-μm sections were stained with hematoxylin and eosin, Masson’s trichrome or anti-CD206 and anti-CD16 antibodies. The wall thickness and infiltration of inflammatory cells were analyzed in small coronary vessels.
Hypoxic stimulation
HUVECs were placed in the hypoxic chamber with the AnaeroPack and a disposable oxygen-absorbing and CO2-generating agent and incubated at 37 °C for the indicated time. The control cells were incubated at 37 °C in an atmosphere of 21% O2 and 5% CO2 for the same duration as the hypoxic cells. The AnaeroPack started to absorb oxygen within 1 min; oxygen tension inside the box dropped to 1 mm Hg within 1 h (O2 <1%, CO2 approximately 5%) and continued to decrease for 24 h.
RNA interference
HUVECs were transfected with HIF-1α-annealed siGENOME SMART pool. We used an siCONTROL Non-Targeting siRNA no. 1 as a negative control. Cells were transfected by small interfering RNA (siRNA) (final concentration 100 nM) at 70–80% confluency using a transfection reagent, DharmaFECT 1 (Dharmacon, Lafayette, CO, USA), in the complete medium according to the manufacturer’s instructions. The medium was changed at 40 h after transfection, and the cells were stimulated by CF6 in the serum-free medium. In general, this procedure downregulated HIF-1α gene expression by 70–80% in HUVECs.
Annexin V and propidium iodide assay for apoptosis
Flow cytometric analysis with an apoptosis detection kit was used to assess apoptotic cells. Briefly, the cells were trypsinized, washed with cold phosphate-buffered saline two times and incubated in the binding buffer (10 mmol l−1 HEPES, 140 mmol l−1 NaCl, 2.5 mmol l−1 CaCl2) with annexin-V-FITC and propidium iodide for 10 min on ice in the dark. The sample was analyzed using a dual-laser FACS VantageSE flow cytometer (Becton Dickinson, Mountain View, CA, USA) within a 1-h period. The percentage of apoptotic cells was estimated for each sample.
Staining for apoptotic cells
The recognition of apoptotic nuclei in paraffin-embedded tissue sections was performed using the ApopMark Apoptosis Detection Kit. Briefly, after permeabilizing the samples and inactivating the endogenous peroxidase, terminal deoxynucleotidyl transferase was added to bind to exposed 3′-OH ends of DNA fragments generated in response to apoptotic signals and to catalyze the addition of biotin-labeled deoxynucleotides. Biotinylated nucleotides were detected using a streptavidin-horseradish peroxidase conjugate and were diaminobenzidine reacted with the horseradish peroxidase-labeled sample to generate an insoluble, colored substrate at the site of DNA fragmentation.
Statistics
All data are shown as the mean±1 s.e.m. A paired or unpaired t-test for the comparison of two variables and a one-way analysis of variance for multiple comparisons followed by Bonferroni’s test were used for statistical analysis. The level of significance was P<0.05.
Results
Effects of CF6 on CXCR4 gene and protein expression
As shown in Figure 1a, the ratio of CXCR4 to GAPDH mRNA by real-time quantitative reverse transcription-PCR was decreased by 65±14% (P<0.05) at 24 h after CF6 treatment at 10−7 M and plateaued at 48 h (69±20%). As shown in Figure 1b, the ratios of CXCR4 to GAPDH mRNA decreased in a dose-dependent manner during the 24-h exposure to CF6 (P<0.05). Figure 1c shows the effects of CF6 at 10−7 M with or without anti-α- and anti-β-subunit antibodies and PP1, a specific c-Src inhibitor, on the ratios of CXCR4 to GAPDH mRNA. Pretreatment with anti-α- and anti-β-subunit antibodies and PP1 inhibited the CF6-induced decrease in CXCR4 gene expression (all P<0.05). Treatment with siRNA for HIF-1α dampened the CF6-induced decrease in CXCR4 gene expression (P<0.05).

Effects of human coupling factor 6 (CF6) on the gene and protein expression of CXC chemokine receptor type 4 (CXCR4) in human umbilical vein endothelial cells (HUVECs). (a) Chronological changes after exposure to CF6 at 10−7 M (n=6). P<0.05 vs. 0 h. (b) Dose-related changes after a 24-h exposure to CF6 at 10−9–10−7 M (n=6). P<0.05 vs. CF6 (−). (c) Effects of CF6 at 10−7 M on the ratio of CXCR4 to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA at 24 h and the effect of various compounds on the CF6-induced decrease in CXCR4 gene expression (n=4–12). P<0.05 vs. control (without agonists). PP1 at 50 μM; αAb: antibody for α-subunit of ATP synthase (dilution of 1:1000); βAb: antibody for β-subunit of ATP synthase (dilution of 1:1000); small interfering RNA (siRNA): after a 40-h treatment with annealing siRNA targeting hypoxia-inducible factor (HIF)-1α at 100 nM. (d) Effect of CF6 and/or hypoxia on the ratio of CXCR4 to GAPDH mRNA at 24 h (n=6). P<0.05 vs. normoxia without CF6. (e) Representative bands for CXCR4 protein and the ratio of CXCR4 to GAPDH in the presence and absence of CF6 at 10−7 M for 24 h (n=4). P<0.05 vs. CF6 (−).
Because hypoxia induces intracellular acidosis and HIF-1α expression, we tested the hypothesis that like CF6, hypoxia may attenuate CXCR4 expression through the same pathway. As shown in Figure 1d, the ratio of CXCR4 to GAPDH mRNA was decreased by both hypoxia and CF6 at 10−7 M to a similar degree (both P<0.05), but no additive effect was found.
Figure 1e illustrates the representative bands for CXCR4 proteins in the presence or absence of CF6 at 10−7 M for 24 h. The immunoreactive band for CXCR4 at 40–47 kDa was decreased after treatment with CF6. The ratio of CXCR4 to GAPDH protein was decreased by 41±10% in HUVECs treated with CF6 at 10−7 M for 24 h (P<0.05).
Mechanism for CF6-induced decrease in CXCR4
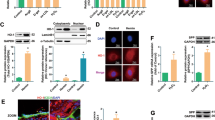
The hypoxia response element (HRE) (GCGTG), which is located at position −29 to −25 relative to the transcription start site, is reported to be involved in acidosis-induced CXCR4 repression in HUVECs.6 To demonstrate whether CF6 regulates endogenous binding of HIF-1α to the CXCR4 promoter, we performed a ChIP assay using primers encompassing the −29 to −25 site and adjacent flanking regions. As shown in Figure 2a, endogenous HIF-1α was bound to the CXCR4 promoter in anti-HIF-1α/chromatin immunoprecipitates, and CF6 increased the amount of HIF-1α bound to the CXCR4 promoter.

Mechanism of coupling factor 6 (CF6)-induced decrease in CXC chemokine receptor type 4 (CXCR4) in human umbilical vein endothelial cells (HUVECs). (a) Chromatin immunoprecipitation (ChIP) assay performed with primers encompassing the hypoxia response element (HRE) in the presence or absence of CF6 at 10−7 M for 7 h. An anti-hypoxia-inducible factor (HIF)-1α antibody (dilution of 1:40) was used to immunoprecipitate the associated chromatin. (b) Histone deacetylase 3 (HDAC3) in immunoprecipitates using an anti-phospho-c-Src (pcSrc) antibody after a 30-min exposure of HUVECs to CF6 at 10−7 M (n=4). PcSrc after a 30-min exposure to CF6 at 10−7 M (n=4). HDAC3 and PcSrc were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the sample before immunoprecipitation. P<0.05 vs. CF6 (−). (c) ChIP assay performed with primers encompassing the HRE in the presence or absence of CF6 at 10−7 M for 7 h. An anti-HDAC3 antibody (dilution of 1:40) was used to immunoprecipitate the associated chromatin.
Because protein-to-protein interaction between c-Src and HDAC3 has been reported to be responsible for CXCR4 repression,12 we examined the possible interaction between phospho-c-Src and HDAC3 using an immunoprecipitation technique. As shown in Figure 2b, HDAC3 association with phospho-c-Src was increased by CF6 at 30 min (left panel). c-Src was also phosphorylated by CF6 at 30 min (right panel). A ChIP assay using primers encompassing HRE at the −29 to −25 site was performed under stimulation with CF6 using anti-HDAC3 antibody. Endogenous binding of HDAC3 to HRE was not affected by CF6 at 30 min (Figure 2c), suggesting that HRE is specific to the CF6-induced HIF-1α pathway but not to the c-Src pathway.
Effect of CF6 on hypoxia-induced apoptosis in HUVECs
The percentage of apoptotic cells was slightly but significantly increased from 4.3±0.7 to 5.6±1.7% by CF6 at 10−7 M in normoxia over 24 h; the same percentage was increased from 5.3±1.8 to 7.0±1.3% in hypoxia (both P<0.05). As shown in Figure 3a, the increase in apoptotic cells by CF6 in both normoxia and hypoxia was blocked by siRNA for HIF-1α. Administration of the CXCR4 ligand, SDF-1, also dampened the CF6-induced increase in apoptotic cells in both normoxia and hypoxia (Figure 3b).

Flow cytometric analysis of apoptosis with annexin-V-FITC and propidium iodide (PI) label in human umbilical vein endothelial cells (HUVECs). (a) Representative charts under normoxia and hypoxia for 24 h with or without CF6 at 10−7 M and a 40-h pretreatment with HIF-1α small interfering RNA (siRNA) at 100 nM. P<0.05 vs. control (n=3). (b) Representative charts under normoxia and hypoxia for 24 h with or without CF6 and stromal-derived factor-1α (SDF-1α) at 300 ng ml−1. P<0.05 vs. control (n=3). A full color version of this figure is available at the Hypertension Research journal online.
CXCR4 protein expression and histological analysis in TG heart
Figure 4a illustrates the representative bands for CXCR4 proteins in the hearts of TG and WT at 50 weeks of age. The immunoreactive band for CXCR4 at 40–47 kDa was decreased in the TG hearts compared with the WT hearts, and the ratio of CXCR4 to GAPDH protein was decreased by 25±4% in the TG hearts (P<0.05). Figures 4b and c (50 weeks of age) and Figures 4e and f (20–25 weeks of age) illustrate the representative Masson’s trichrome staining and immunostaining for CD16 and CD206 in the small coronary arteries and perivascular tissues obtained from the TG and WT hearts. The increase in wall thickness and infiltration of CD16- or CD206-positive cells in small coronary arteries and perivascular tissues was observed in the TG at the age of 50 weeks (all P<0.05) but not in the TG at the age of 20–25 weeks. The number of apoptotic cells in the coronary small arteries was greater in the TG than in the WT (Figure 4d).

Wall thickness and inflammation in coronary artery obtained from transgenic mice (TG) overexpressing coupling factor 6 (CF6) at the age of 20–15 weeks and 50 weeks. (a) Representative bands for CXCR4 in the heart of wild-type (WT) mice and TG at 50 weeks of age. P<0.05 vs. WT (n=4). (b) Representative Masson’s trichrome staining in the small coronary arteries of WT and TG. Inside diameter and wall thickness of WT and TG at 50 weeks of age. P<0.05 vs. WT (n=4). (c) Representative immunostaining for CD16 and CD206 in the small coronary arteries of WT and TG at 50 weeks of age. Solid column: CD16-positive cells; open column: CD206-positive cells. P<0.05 vs. WT (n=5). (d) Representative staining of apoptotic cells in the small coronary arteries of WT and TG at 50 weeks of age. P<0.05 vs. WT (n=4). (e) Representative Masson’s trichrome staining in the small coronary arteries of WT and TG. Inside diameter and wall thickness of WT and TG at 20–25 weeks of age (n=4). (f) Representative immunostaining for CD16 and CD206 in the small coronary arteries of WT and TG at 20–25 weeks of age. Solid column: CD16-positive cells; open column: CD206-positive cells (n=4). A full color version of this figure is available at the Hypertension Research journal online.
Discussion
The present study aimed to investigate whether CF6 regulates CXCR4 expression and function in vascular endothelial cells and to gain insights into the molecular mechanisms and in vivo role of CF6. The results showed that CF6 decreased the gene and protein expression of CXCR4 by enhancing endogenous HIF-1α binding to the HRE of the CXCR4 promoter and the interaction between phospho-c-Src and HDAC3. Apoptotic cells were increased at 24 h by CF6 in normoxia and hypoxia; however, this effect was abolished by HIF-1α knockdown. Wall thickness and infiltration of inflammatory cells were increased in the coronary small arteries and perivascular tissues obtained from TG.
Effect of CF6 and hypoxia on CXCR4 expression
CF6 stimulates ATPase activity at the surface of HUVECs after binding to the β-subunit of plasma membrane ATP synthase and induces intracellular acidosis by a flux of hydrogen ions through Fo.7 The present results showed that CF6 decreased the gene and protein expression of CXCR4 in vascular endothelial cells, and the expression was attenuated by anti-β-subunit antibody. Because anti-β-subunit antibody blocks CF6-induced acidification by blocking CF6’s receptor, the decrease in CXCR4 in response to CF6 may be associated with intracellular pH. This notion is supported by a previous report that direct administration of acid into the extracellular fluid decreased CXCR4 expression in HUVECs.6 Acidosis occurs in a variety of pathological conditions, including ischemia and/or hypoxia, which are known to stimulate HIF-1α. Thus, we examined the effect of hypoxia on CXCR4 expression and the role of HIF-1α in this change. Like CF6, hypoxia decreased the gene expression of CXCR4, but an additive effect on CXCR4 repression in response to CF6 was not found as expected. Either CF6 or hypoxia appeared to influence vascular function via the same signaling as acidosis and HIF-1α, and the CF6-induced CXCR4 reduction was abolished by both anti-HIF-1α antibody and siRNA for HIF-1α.
CF6 decreased the gene and protein expression of CXCR4, but the decrease was only significant at a concentration of more than 10−7 M. Like other vasoactive substances, the plasma CF6 level (10−9–10−8 M) was lower than the critical concentration (10−7 M) at which CXCR4 expression in cultured vascular endothelial cells was decreased in vitro.13, 14 We previously showed that intravenous administration of anti-CF6 antibody to rats counteracted the biological effects of CF6.15 Therefore, CF6 is active in vivo at a lower concentration compared with the concentration that was effective in vitro.
Regulation mechanism for CXCR4 gene expression
We next investigated the molecular mechanisms for CF6-induced CXCR4 repression by analyzing the binding of transcription factors to the CXCR4 promoter and the protein-to-protein interaction with this promoter. HIF-1α, which is activated by acidosis,16, 17, 18, 19 binds to the HRE of the CXCR4 promoter.20, 21 The present ChIP assay demonstrated that HIF-1α bound to the CXCR4 promoter at HRE position −29 to −25 after HUVECs were treated with CF6. Although HIF-1α acts as a transcriptional activator or repressor in a number of promoters,22, 23 the CXCR4 promoter at HRE position −29 to −25 has been reported to be associated with CXCR4 repression.6 Thus, one mechanism for CF6-induced CXCR4 repression is the binding of HIF-1α to the HRE at position −29 to −25 of the CXCR4 promoter.
Recently, it was reported that tyrosine kinase c-Src interacts at the membrane level with the substrate HDAC312 and regulates CXCR4 gene transcription. Furthermore, the complex formed by c-Src and HDAC3 was shown to downregulate nuclear factor-κB and Ets1 activities, which are responsible for CXCR4 repression.24 Using PP1 as a specific c-Src inhibitor and anti-α- and anti-β-subunit antibodies as CF6 receptor blockers, we examined whether CF6 receptor-mediated c-Src activation contributes to CXCR4 repression in HUVECs.25 Anti-α- and anti-β-subunit antibodies or PP1 attenuated the CF6-induced decrease in CXCR4, suggesting that CXCR4 repression depends on CF6 receptor-mediated c-Src activation. We further investigated the interaction between c-Src and HDAC3 using the immunoprecipitation technique and showed that HDAC3 was bound with phospho-c-Src at 30 min after CF6 treatment in HUVECs. Because endogenous binding of HDAC3 to the HRE was unaffected by CF6, the interaction between c-Src and HDAC3 appeared to repress CXCR4 mRNA independently of the HIF-1α-mediated mechanism.
Effect of CF6 on cell death
CXCR4 is a key receptor that regulates a number of downstream effectors related to endothelial function. CXCL12 binding to CXCR4 promotes the activation of multiple G-protein-dependent signaling pathways, resulting in diverse biological responses, such as migration, adhesion, survival and/or proliferation and transcription activation. Of these responses, CXCR4 activation by its chemokine, SDF-1, may lead to the simultaneous activation of both anti- and proapoptotic signaling pathways; the balance between these pathways ultimately influences cell survival. The antiapoptotic second messenger signal is the Giα-protein-dependent activation of Akt and extracellular signal-regulated protein kinase, whereas the proapoptotic signaling pathway is the Giα-protein-independent activation of p38 MAP kinase.26 In the present study, we showed that apoptotic cells were slightly but significantly increased by CF6 treatment at 24 h in normoxia and hypoxia, and this increase was abolished by pretreatment with either siRNA for HIF-1α or CXCR4 ligand. Because acidosis directly activates p38 MAP kinase,16 CF6 may upregulate the activity of p38 MAP kinase as proapoptotic signaling. Concerning antiapoptotic signaling, we previously showed that CF6 downregulates Akt phosphorylation in the heart.27 Thus, it appears that the SDF-1/CXCR4 axis exerts an antiapoptotic signal in HUVEC, whereas CF6 induces apoptosis via downregulation of CXCR4 signaling. Overall, these observations suggest that CF6 may function as a proapoptotic molecule through HIF-1α-mediated suppression of the SDF-1/CXCR4 axis.
To further investigate the in vivo role of CF6’s proapoptotic effect, we analyzed histological changes in TG-derived coronary small arteries. Endothelial dysfunction is an initial step for arteriosclerosis, and inflammation is closely related to arteriosclerosis progression. In the present study, an increase in wall thickness, infiltration of CD16-positive cells (inflammatory macrophages), CD206-positive cells (fibrosis-promoting macrophages) and apoptotic cells were observed in coronary small arteries and perivascular tissues obtained from 50-week-old TG. Thus, the proapoptotic signaling of CF6 may be involved in intimal thickness and inflammatory cell infiltration in vivo.
Implications of linkage between CF6 and CXCR4 in cardiovascular disorders
In clinical settings, we and others have shown that circulating CF6 is elevated in patients with hypertension, acute myocardial infarction, end-stage renal disease, stroke and diabetes,13, 14, 28, 29, 30, 31 all of which are predispositions to arteriosclerosis. The present study showed that CF6 is involved in endothelial dysfunction and vascular impairment by downregulating CXCR4 signaling. Given the present finding and the widespread biological actions of CF6, such as inhibition of prostacyclin and NO,8, 9 reduction in CF6 levels may be important and useful to prevent arteriosclerosis and eventually cardiovascular events. To date, we have reported that salt restriction, vitamin C and vitamin B12 plus folic acid lower the plasma CF6 levels of patients with hypertension and stroke13, 14 and that peroxisome proliferator-activated receptor γ ligand attenuate CF6 release from cultured vascular endothelial cells.32 Thus, the present findings may provide new insights into our understanding of the pivotal role played by CF6 in the mechanism underlying endothelial dysfunction.
In conclusion, this report first showed that CF6 decreased CXCR4 expression through HIF-1α- and c-Src-mediated pathways in vascular endothelial cells. Because CXCR4 has an important role in endothelial survival function, CF6 may be involved in the impairment of vascular homeostasis under physiological and pathological conditions. In light of the current findings, it appears that CF6 has an important role in the progression of arteriosclerosis by complex mechanisms.
References
Teicher BA, Fricker SP . CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 2010; 16: 2927–2931.
Shen X, Artinyan A, Jackson D, Thomas RM, Lowy AM, Kim J . Chemokine receptor CXCR4 enhances proliferation in pancreatic cancer cells through AKT and ERK dependent pathways. Pancreas 2010; 39: 81–87.
Busillo JM, Benovic JL . Regulation of CXCR4 signaling. Biochim Biophys Acta 2007; 1768: 952–963.
Ahr B, Denizot M, Robert-Hebmann V, Brelot A, Biard-Piechaczyk M . Identification of the cytoplasmic domains of CXCR4 involved in Jak2 and STAT3 phosphorylation. J Biol Chem 2005; 280: 6692–6700.
Ziegelstein RC, Cheng L, Capogrossi MC . Flow-dependent cytosolic acidification of vascular endothelial cells. Science 1992; 258: 656–659.
Melchionna R, Romani M, Ambrosino V, D'Arcangelo D, Cencioni C, Porcelli D, Toietta G, Truffa S, Gaetano C, Mangoni A, Pozzoli O, Cappuzzello C, Capogrossi MC, Napolitano M . Role of HIF-1alpha in proton-mediated CXCR4 down-regulation in endothelial cells. Cardiovasc Res 2010; 86: 293–301.
Osanai T, Magota K, Tanaka M, Shimada M, Murakami R, Sasaki S, Tomita H, Maeda N, Okumura K . Intracellular signaling for vasoconstrictor coupling factor 6: novel function of β-subunit of ATP synthase as receptor. Hypertension 2005; 46: 1140–1146.
Osanai T, Kamada T, Fujiwara N, Katoh T, Takahashi K, Kimura M, Satoh K, Magota K, Kodama S, Tanaka T, Okumura K . A novel inhibitory effect on prostacyclin synthesis of coupling factor 6 extracted from the heart of spontaneously hypertensive rats. J Biol Chem 1998; 273: 31778–31783.
Tanaka M, Osanai T, Murakami R, Sasaki S, Tomita H, Maeda N, Satoh K, Magota K, Okumura K . Effect of vasoconstrictor coupling factor 6 on gene expression profile in human vascular endothelial cells: enhanced release of asymmetric dimethylarginine. J Hypertens 2006; 24: 489–497.
Osanai T, Tomita H, Kushibiki M, Yamada M, Tanaka M, Ashitate T, Echizen T, Katoh C, Magota K, Okumura K . Coupling factor 6 enhances Src-mediated responsiveness to angiotensin II in resistance arterioles and cells. Cardiovasc Res 2009; 81: 780–787.
Osanai T, Tanaka M, Magota K, Tomita H, Okumura K . Coupling factor 6-induced activation of ecto-F1Fo complex induces insulin resistance, mild glucose intolerance and elevated blood pressure in mice. Diabetologia 2012; 55: 520–529.
Longworth MS, Laimins LA . Histone deacetylase 3 localizes to the plasma membrane and is substrate of Src. Oncogene 2006; 25: 4495–4500.
Osanai T, Sasaki S, Kamada T, Fujiwara N, Nakano T, Tomita H, Matsunaga T, Magota K, Okumura K . Circulating coupling factor 6 in human hypertension: role of reactive oxygen species. J Hypertens 2003; 21: 2323–2328.
Osanai T, Fujiwara N, Sasaki S, Metoki N, Saitoh G, Tomita H, Nishimura T, Shibitani S, Yokoyama H, Konta Y, Magota K, Okumura K . Novel pro-atherogenic molecule coupling factor 6 is elevated in patients with stroke: a possible linkage to homocysteine. Ann Med 2010; 42: 79–86.
Osanai T, Tanaka M, Kamada T, Nakano T, Takahashi K, Okada S, Sirato K, Magota K, Kodama S, Okumura K . Mitochondrial coupling factor 6 as a potent endogenous vasoconstrictor. J Clin Invest 2001; 108: 1023–1030.
Zheng M, Reynolds C, Jo SH, Wersto R, Han Q, Xiao RP . Intracellular acidosis-activated p38 MAPK signaling and its essential role in cardiomyocyte hypoxic injury. FASEB J 2005; 19: 109–111.
Ihnatko R, Kubes M, Takacova M, Sedlakova O, Sedlak J, Pastorek J, Kopacek J, Pastorekova S . Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol 2006; 29: 1025–1033.
Willam C, Warnecke C, Schefold JC, Kugler J, Koehne P, Frei U, Wiesener M, Eckardt KU . Inconsistent effects of acidosis on HIF-alpha protein and its target genes. Pflugers Arch 2006; 451: 534–543.
Xu J, Peng Z, Li R, Dou T, Xu W, Gu G, Liu Y, Kang Z, Tao H, Zhang JH, Ostrowski RP, Lu J, Sun X . Normoxic induction of cerebral HIF-1alpha by acetazolamide in rats: role of acidosis. Neurosci Lett 2009; 451: 274–278.
Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A . Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 2003; 198: 1391–1402.
Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W . Chemokine receptor CXCR4 downregulated by von Hippel–Lindau tumour suppressor pVHL. Nature 2003; 425: 307–311.
Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL . Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005; 105: 659–669.
Wenger RH, Stiehl DP, Camenisch G . Integration of oxygen signaling at the consensus HRE. Sci STKE 2005; 2005: re12.
Matteucci E, Ridolfi E, Maroni P, Bendinelli P, Desiderio MA . c-Src/histone deacetylase 3 interaction is crucial for hepatocyte growth factor dependent decrease of CXCR4 expression in highly invasive breast tumor cells. Mol Cancer Res 2007; 5: 833–845.
Kumagai A, Osanai T, Katoh C, Tanaka M, Tomita H, Morimoto T, Murakami R, Magota K, Okumura K . Coupling factor 6 downregulates platelet endothelial cell adhesion molecule-1 via c-Src activation and acts as a proatherogenic molecule. Atherosclerosis 2008; 200: 45–50.
Vlahakis SR, Villasis-Keever A, Gomez T, Vanegas M, Vlahakis N, Paya CV . G protein-coupled chemokine receptors induce both survival and apoptotic signaling pathways. J Immunol 2002; 169: 5546–5554.
Sagara S, Osanai T, Itoh T, Izumiyama K, Shibutani S, Hanada K, Yokoyama H, Yamamoto Y, Yokota T, Tomita H, Magota K, Okumura K . Overexpression of coupling factor 6 attenuates exercise-induced physiological cardiac hypertrophy by inhibiting PI3K/Akt signaling in mice. J Hypertens 2012; 30: 778–786.
Li XL, Xing QC, Gao YY, Dong B, Pang YZ, Jiang HF, Tang CS . Plasma level of mitochondrial coupling factor 6 increases in patients with type 2 diabetes mellitus. Int J Cardiol 2007; 117: 411–412.
Osanai T, Nakamura M, Sasaki S, Tomita H, Saitoh M, Osawa H, Yamabe H, Murakami S, Magota K, Okumura K . Plasma concentration of coupling factor 6 and cardiovascular events in patients with end-stage renal disease. Kidney Int 2003; 64: 2291–2297.
Ding WH, Chu SY, Jiang HF, Cai DY, Pang YZ, Tang CS, Qi YF . Plasma mitochondrial coupling factor 6 in patients with acute myocardial infarction. Hypertens Res 2004; 27: 717–722.
Chai SB, Hui YM, Li XM, Tang CS . Plasma level of mitochondrial coupling factor 6 increases in patients with coronary heart disease. Circ J 2007; 71: 693–697.
Tomita H, Osanai T, Toki T, Sasaki S, Maeda N, Murakami R, Magota K, Yasujima M, Okumura K . Troglitazone and 15-deoxy-deta 12, 14-prostaglandin J2 inhibit shear-induced coupling factor 6 release in endothelial cells. Cardiovasc Res 2005; 67: 134–141.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (No. 19590800 for Tomohiro Osanai) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Suzuki, A., Osanai, T., Tanaka, M. et al. Coupling factor 6 attenuates CXCR4 expression through the HIF-1α and c-Src pathways and promotes endothelial apoptosis and inflammation. Hypertens Res 37, 708–715 (2014). https://doi.org/10.1038/hr.2014.65
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2014.65
Keywords
This article is cited by
-
Sirtuin 6 attenuates angiotensin II-induced vascular adventitial aging in rat aortae by suppressing the NF-κB pathway
Hypertension Research (2021)
-
Lung-specific RNA interference of coupling factor 6, a novel peptide, attenuates pulmonary arterial hypertension in rats
Respiratory Research (2016)
-
An exaggerated blood pressure response to exercise is associated with nitric oxide bioavailability and inflammatory markers in normotensive females
Hypertension Research (2016)
