
Abstract
Azilsartan is a novel angiotensin receptor blocker being developed for hypertension treatment. This 16-week, multicenter, randomized, double-blind study compared the efficacy and safety of azilsartan (20–40 mg once daily by forced titration) and its ability to provide 24-h blood pressure (BP) control, with that of candesartan cilexetil (candesartan; 8–12 mg once daily by forced titration) in 622 Japanese patients with grade I–II essential hypertension. Efficacy was evaluated by clinic-measured sitting BP, and by ambulatory BP monitoring (ABPM) at week 14. Participants (mean age: 57 years, 61% males) had a mean baseline sitting BP of 159.8/100.4 mm Hg. The mean change from baseline in sitting diastolic BP at week 16 (primary endpoint) was −12.4 mm Hg in the azilsartan group and −9.8 mm Hg in the candesartan group, demonstrating a statistically significant greater reduction with azilsartan vs. candesartan (difference: −2.6 mm Hg, 95% confidence interval (CI): −4.08 to −1.22 mm Hg, P=0.0003). The week 16 (secondary endpoint) mean change from baseline in sitting systolic BP was −21.8 mm Hg and −17.5 mm Hg, respectively, a significant decrease with azilsartan vs. candesartan (difference: −4.4 mm Hg, 95% CI: −6.53 to −2.20 mm Hg, P<0.0001). On ABPM, the week 14 mean changes from baseline in diastolic and systolic BP were also significantly greater with azilsartan over a 24-h period, and during the daytime, night-time and early morning. Safety and tolerability were similar among the two groups. These data demonstrate that once-daily azilsartan provides a more potent 24-h sustained antihypertensive effect than that of candesartan but with equivalent safety.
Similar content being viewed by others

Introduction
Hypertension is a primary risk factor for cardiovascular disease, and strict blood pressure (BP) control is critical from the standpoint of prevention of cardiovascular disease.1 In Japan, where the number of people with hypertension is approximately 40 million, it is estimated that around 50% of patients on antihypertensive treatment are inadequately managed in terms of their office BP, highlighting the need for stricter BP control to prevent adverse sequelae.2, 3 Strict BP control over 24 h, including the night-time and early morning hours, is crucial, as the average BP level over 24 h, nocturnal and early morning BP are more closely associated with target organ damage and cardiovascular events than office BP.4, 5, 6 In clinical practice, however, it is not infrequent that existing antihypertensive drugs fail to provide an adequate antihypertensive effect that is sustained over 24 h.7 In the J-MORE (Jichi Morning Hypertension Research) study, 60.7% of treated hypertensives with well-controlled clinic BP were found to have masked morning hypertension (systolic BP (SBP) ⩾135 mm Hg; diastolic BP (DBP) ⩾85 mm Hg), and only 16.4% of patients overall had well-controlled clinic and morning BP levels.8 These findings may be related to the limited BP-lowering effect and duration of action of some antihypertensives, highlighting the importance of treatments that control BP over a full 24-h period when administered once daily.
The current Japanese Society of Hypertension Guidelines for the Management of Hypertension2 advocate the use of five classes of drugs as first-line treatments for hypertension: calcium channel blockers, angiotensin II (AII) receptor blockers (ARBs), angiotensin-converting enzyme (ACE) inhibitors, diuretics and β-adrenoceptor blockers (β-blockers). Among the recommended first-line agents, the ARBs are now widely used as a key component of antihypertensive regimens because of their favorable efficacy/tolerability profiles.9 In addition, clinical outcome trials have shown that the ARBs reduce the proportion of hypertensive patients who develop type 2 diabetes mellitus,10 and improve cardiovascular outcomes in such conditions as high-risk hypertension,11, 12 heart failure13, 14 and diabetic kidney disease.15, 16
Azilsartan is a new ARB that inhibits the binding of AII to AII type 1 (AT1) receptors selectively, and is thus expected to exert a more potent and sustained BP-lowering effect than existing ARBs. In an in vitro study, azilsartan was shown to have a higher affinity for and slower dissociation from AT1 receptors than other ARBs (olmesartan, telmisartan, valsartan and irbesartan).17 The present study was designed to compare the efficacy and tolerability of azilsartan 20–40 mg once daily with candesartan cilexetil (the most commonly used ARB in Japan) 8–12 mg once daily (the most frequently used dosage and maximum clinically approved dosage in Japan) in patients with grade I or II essential hypertension. The study also evaluated the ability of these drugs to provide 24-h BP control using ambulatory BP monitoring (ABPM).
Methods
Study design
This was a phase III, randomized, double-blind, parallel-group study comparing the efficacy and safety of azilsartan and candesartan cilexetil (candesartan) in Japanese patients with grade I or II essential hypertension. In addition, the persistence of the effect of azilsartan, measured by ABPM, was assessed. The study was conducted at 33 centers in Japan between May 2009 and June 2010. After a 4-week single-blind, placebo run-in period, eligible patients were randomized equally (via an interactive web response system) to receive either azilsartan or candesartan in a double-blinded manner using plasma renin activity (<0.5 ng ml−1 per h or ⩾0.5 ng ml−1 per h) at week −2 as a stratification factor for randomization. During the 16-week, double-blind treatment period, all patients in each group received the assigned study drug once daily before or after breakfast. Patients in the azilsartan group received a dosage of 20 mg daily for the first 8 weeks and then 40 mg daily for the subsequent 8 weeks. Patients in the candesartan group received a dosage of 8 mg daily for the first 8 weeks and then 12 mg daily for the subsequent 8 weeks.
The study was approved by Institutional Review Boards at each study center, and was conducted in accordance with the ethical provisions set out in the Declaration of Helsinki, the International Conference on Harmonisation, Harmonised Tripartite Guideline for GCP (Good Clinical Practice) E6(R1) and all applicable local laws and regulations. All patients were required to provide written informed consent prior to the initiation of any study-related procedures.
The trial was registered with the Japan Pharmaceutical Information Center Clinical Trials Information (JapicCTI) at http://www.clinicaltrials.jp/user/cte_main_e.jsp (identifier JapicCTI-090762).
Patients
Patients with a diagnosis of grade I or II essential hypertension were assessed during the single-blind, placebo run-in period. To be eligible for the study, patients were required to be ⩾20 years of age and have a sitting DBP of ⩾95 and <110 mm Hg, and a sitting SBP of ⩾150 and <180 mm Hg at weeks −2 and 0 of the run-in period. Women of child-bearing potential were required to practice effective contraception during the course of the study. Exclusion criteria were grade III hypertension (BP ⩾180/⩾110 mm Hg), secondary or malignant hypertension; the presence of cardiovascular disease; a >8 mm Hg decrease in DBP from week −2 to week 0; significant hepatic or renal disease; hyperkalemia; malignancy; known hypersensitivity to ARBs, ACE inhibitors or direct renin inhibitors; a history of drug or alcohol abuse within the previous 2 years; a day–night reversal lifestyle; and pregnancy or lactation. Other medications not permitted during the study period were antihypertensive and antianginal drugs, hypnosedative/antianxiety agents, antidepressants, non-steroidal anti-inflammatory drugs, glucocorticoids, liquorice preparations, antiarrhythmic drugs, estrogens, digitalis preparations and potassium supplements.
Study procedures
Patients were assessed via measurements of sitting BP and pulse rate and via physical examinations at clinic visits every 2 weeks during both the 4-week placebo run-in period and the 16-week treatment period. Sitting BP was measured by the investigator at least three times at 1- or 2-minute intervals at trough (24±3 h post-dose) using a digital or manual BP monitor, and the mean value of two stable consecutive BP measurements was used for analysis. ABPM, in which BP was measured at 30-minute intervals for ⩾26 h using an oscillometric monitor (TM- 2431, A&D), was undertaken at baseline (week 0) and at week 14, starting at 1000 hours (±1 h). Patients took the study drug ⩾1 h after the start of the measurements in the morning, and after the completion of measurements on the following day. During the period of ABPM, patients were instructed to avoid taking a bath, taking an afternoon nap, performing exercise and consuming alcohol- and caffeine-containing food/drinks. The major quality criteria used for an acceptable ABPM recording included the following: (1) minimum of 80% of the BP readings expected during the 24-h period; (2) no more than 2 nonconsecutive hours with <1 valid BP reading; and (3) no behaviors seriously affecting BP (afternoon nap, drinking and so on).
With regard to adverse events, all patients were queried at every visit with non-leading questions. In addition, a resting 12-lead electrocardiogram was performed at baseline and week 16. Clinical laboratory tests (hematology, serum chemistry, urinalysis) were performed at weeks −4, 0 (baseline), 2, 8 and 16 after the patients had fasted for ⩾10 h.
Outcome criteria
The primary efficacy endpoint was the change from baseline in the sitting trough (pre-dose) DBP at the end of the treatment period (week 16), using last observation carried forward (LOCF) data analysis. Secondary efficacy endpoints included: (1) the changes from baseline to week 8 (before dose escalation) in sitting trough DBP (LOCF analysis) and from baseline to weeks 8 (before dose escalation) and 16 in sitting trough SBP (LOCF analysis); (2) sitting trough SBP and DBP at each scheduled 2-week measurement time point; (3) the proportions of responders (⩾20 mm Hg decrease in sitting trough SBP and ⩾10 mm Hg decrease in sitting trough DBP, or a sitting trough SBP of <130 mm Hg and a sitting trough DBP of <85 mm Hg) and well-controlled patients (sitting trough SBP <130 mm Hg and sitting trough DBP <85 mm Hg); (4) mean changes in DBP and SBP on ABPM over a 24-h period, during both the daytime (while awake) and night-time (while asleep). Safety was evaluated via the occurrence of adverse events, clinical laboratory tests, vital signs, body weight and resting 12-lead electrocardiogram findings.
Statistical analysis
Sample size
The sample size was determined on the basis of the results of an earlier dose-ranging study in Japanese patients (unpublished data; Takeda Pharmaceutical Company). The difference in the mean change of sitting trough DBP from baseline to the end of the treatment period between azilsartan 40 mg and candesartan 12 mg (azilsartan group − candesartan group) was assumed to be −2.5 mm Hg, with a s.d. common to all groups of 11.0 mm Hg. A sample size of 305 subjects per group was required to verify the statistical difference between azilsartan 40 mg and candesartan 12 mg with at least an 80% power and a two-sided significance level of 5%. Accordingly, the number of subjects evaluable for the primary endpoint was determined to be 305 per group, or 610 in total.
Analysis of endpoints
The efficacy analysis was performed in the full analysis set, which included patients who were randomized and received the study medications at least once. Safety was analyzed in the safety analysis set, which included patients who received study medication at least once.
For the primary efficacy endpoint, summary statistics and two-sided 95% confidence intervals (CI) of the mean values were determined and a one-sample t-test was performed. The point estimate of the least-square (LS) mean and its two-sided 95% CI for each treatment group were calculated using a two-way analysis of variance model with the change from baseline (week 0) in the sitting trough DBP at week 16 (LOCF data) as a dependent variable, and the plasma renin activity (<0.5 ng ml−1 per h or ⩾0.5 ng ml−1 per h) at week −2 and treatment group as independent variables. The same model was used to calculate the point estimate of the difference in LS means between the azilsartan and candesartan groups and its two-sided 95% CI.
The minimum clinically significant difference in the sitting trough DBP between the two drugs in this study was set at 1.6 mm Hg as this value was one-third of the placebo-adjusted treatment effect of candesartan 8 mg on sitting trough DBP in the dose-ranging study in Japanese patients (unpublished data; Takeda Pharmaceutical Company), and because the results of the secondary efficacy endpoint in the present study (the change in the sitting trough DBP at week 8 of treatment) were interpreted the same way as for the primary endpoint. Azilsartan was regarded as non-inferior to candesartan if the upper limit of the two-sided 95% CI of the difference in the estimated LS means between the azilsartan and candesartan groups was below 1.6 mm Hg; and superior to candesartan if the upper limit of the two-sided 95% CI was below 1.6 mm Hg. The difference between azilsartan and candesartan was regarded as statistically significant if the upper limit of the two-sided 95% CI was below zero.
Secondary efficacy endpoints were analyzed similarly to the primary efficacy endpoint except for the proportions of responders and well-controlled patients, the data for which were summarized by treatment group and compared at each time point using the Cochrane-Mantel-Haenszel test with the plasma renin activity at week −2 as a stratification factor. For the ABPM endpoints, that is, the changes from baseline in the mean DBP, SBP and pulse rate over 24 h (defined as midday on day 1 of measurement to 1159 hours on day 2), during the daytime/waking hours (midday on day 1 to 1 min before bedtime and from wake-up time to 1159 hours on day 2), and at night-time (bedtime to 1 min before waking), summary statistics and two-sided 95% CIs of the mean values were presented by treatment group and by period and a one-sample t-test was performed. ANCOVA was also performed using the plasma renin activity (<0.5 ng ml−1 per h or ⩾0.5 ng ml−1 per h) at week −2 and the treatment group as independent variables, and baseline values as covariates.
Results
Patients disposition and demographics
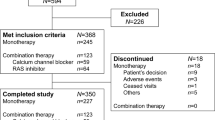
A total of 1022 patients provided informed consent for participation in the trial, of whom 636 were randomized, 319 to azilsartan and 317 to candesartan (Figure 1); however, of the 636 randomized patients, 1 patient in the candesartan group did not receive treatment because of a GCP violation at the study site. Of the 635 patients who were treated, 587 (92.4%) completed the study (azilsartan 293; candesartan 294). The most common reasons for withdrawal were lack of efficacy, pretreatment event/adverse event and voluntary withdrawal (Figure 1). There was no difference between the treatment groups in the percentage of patients who were withdrawn.

Disposition of patients in the study.
Due to a major violation of GCP (conducted by a non-contracted physician), at one of the study sites, which resulted in the exclusion of 6 patients who received azilsartan and 7 who received candesartan, the full analysis set comprised 622 patients, 313 of whom received azilsartan and 309 candesartan.
There were no remarkable differences between the treatment groups at baseline for any demographic characteristic (Table 1). The mean age of patients was 57 years in both treatment groups; males accounted for 59% of the azilsartan group and 63% of the candesartan group. The baseline mean sitting and 24-h BPs were 159.8/100.4 mm Hg and 155.0/94.6 mm Hg, respectively, and the mean plasma renin activity at week 2 was 0.6 ng ml−1 per h in both groups. Treatment compliance was between 90% and 100% in ⩾99% of patients in both groups.
Changes in sitting BP levels
For the primary efficacy parameter, the mean change from baseline in sitting trough DBP at week 16 in the full analysis set, both azilsartan and candesartan produced significant decreases (azilsartan: −12.4±9.87 mm Hg, candesartan: −9.8±8.50 mm Hg). The difference in the LS means between the azilsartan and the candesartan groups was −2.6 mm Hg (95% CI −4.08, −1.22). The upper limit of the 95% CI fell below zero, meaning that the decrease was statistically significantly greater in the azilsartan group than in the candesartan group (P=0.0003) (Figure 2). For the secondary efficacy variable, the mean change in sitting trough SBP at week 16 was −21.8 mm Hg and −17.5 mm Hg in the azilsartan and candesartan groups, respectively, resulting in a significant decrease in the azilsartan group compared with the candesartan group (LS mean difference −4.4 mm Hg, 95% CI −6.53, −2.20; P<0.0001). Similarly, the mean changes in sitting trough DBP and in sitting trough SBP at week 8 (before dose escalation) were significantly greater with azilsartan than with candesartan for each variable (Figure 2).

Changes (least-square (LS) means) in sitting trough diastolic blood pressure (DBP) and systolic blood pressure (SBP) from baseline (week 0) to weeks 8 (before dose escalation) and 16 in the azilsartan and candesartan treatment groups (full analysis set populations; last observation carried forward (LOCF) analysis). LS mean differences between the groups and 95% confidence intervals (CIs) and P-values for the differences are shown (two-way analysis of variance model using plasma renin activity at week −2 and treatment group as independent variables). *Primary efficacy endpoint.
SBP and DBP at each scheduled measurement time point
With both ARBs, the mean sitting trough DBP and SBP were significantly decreased from baseline at all scheduled time points from week 2 to week 16 (data not shown). Significantly greater reductions in the sitting trough DBP and SBP were recorded in the azilsartan group in comparison with the candesartan group at all measurement time points. The differences in LS means between the azilsartan and candesartan groups ranged from −1.2 mm Hg to −2.8 mm Hg for DBP, and from −2.3 mm Hg to −5.1 mm Hg for SBP. Sitting trough DBP and SBP were significantly decreased from the day of the dose increase at week 8 in the azilsartan group at every scheduled measurement time point.
Proportions of responders and well-controlled patients
As shown in Table 2, the proportions of responders at week 16 (LOCF) and week 8 (LOCF) were significantly higher in the azilsartan group (51.8% and 44.1%, respectively) than in the candesartan group (34.3% and 29.4%, respectively) (P<0.0001 and P=0.0001, respectively). Similarly, the proportions of well-controlled patients at week 16 (LOCF) and week 8 (LOCF) were also significantly higher in the azilsartan group (19.3% and 18.0%, respectively) than in the candesartan group (13.3% and 10.0%, respectively) (P=0.0409 and P=0.0041, respectively).
In addition, the proportions of well-controlled patients who achieved a sitting trough SBP of <140 mm Hg and a sitting trough DBP of <90 mm Hg were 48.6% and 33.3% at week 16 (LOCF), and 38.9% and 29.4% at week 8 (LOCF) in the azilsartan and candesartan groups, respectively.
Evaluations by ABPM
Reductions from baseline to week 14 in mean DBP and SBP during ABPM were generally greater in the azilsartan group than in the candesartan group, as shown by the 24-h profiles for time-matched changes (Figure 3). The mean reductions from baseline to week 14 in the 24-h, daytime and night-time mean DBP and SBP during ABPM were all significantly greater in the azilsartan group than in the candesartan group (Table 2). For mean DBP, the differences in LS means between the azilsartan and candesartan groups were −2.1 mm Hg, 95% CI −3.29, −0.89, P=0.0007 during 24 h; −2.5 mm Hg, 95% CI −3.83, −1.22, P=0.0002 in the daytime; and −1.5 mm Hg, 95% CI −2.92, −0.10, P=0.0364 at night-time. For mean SBP, the differences in LS means were −3.7 mm Hg, 95% CI −5.70, −1.62, P=0.0005 during 24 h; −4.4 mm Hg, 95% CI −6.63, −2.20, P=0.0001 in the daytime; and −2.9 mm Hg, 95% CI −5.22, −0.61, P=0.0133 at night-time. Although the early morning mean SBP (defined as the average SBP during the first 2-h period after awakening) was not a preplanned study endpoint, the decrease in this parameter from baseline to week 14 was also significantly greater in the azilsartan group than in the candesartan group (−4.4 mm Hg, 95% CI −7.31, −1.42, P=0.0038). In addition, trough-to-peak ratios (not a preplanned study endpoint) on ABPM at week 14 were 0.97 and 0.75 for DBP and 0.95 and 0.82 for SBP in the azilsartan and candesartan groups, respectively (Table 2). The trough value was defined as the mean change in BP during 2 h of premedication on day 2; the peak value was defined as the mean change in BP between the time the BP change was maximal (2–10 h after study drug intake on day 1) and just after that.18

24-h profiles of changes in mean diastolic blood pressure (DBP) and systolic blood pressure (SBP) from baseline (week 0) to week 8 in the azilsartan and candesartan treatment groups (ambulatory blood pressure monitoring (ABPM) data; full analysis set populations).
Subgroup analyses
Mean changes from baseline in sitting trough DBP and SBP at week 16 (LOCF) were investigated in the full analysis set subgroups stratified by baseline characteristics, including sitting DBP (week 0), sitting SBP (week 0), gender, body weight, body mass index, age, plasma renin activity, eGFR and the presence or absence of diabetes mellitus. In both treatment groups, the mean decrease from baseline (week 0) in sitting trough DBP and SBP tended to be greater in patients with high plasma renin activity (⩾0.5 ng ml−1 per h) than in subjects with low plasma renin activity (<0.5 ng ml−1 per h). Independently of plasma renin activity, the mean decrease from baseline (week 0) in sitting trough DBP and SBP at week 16 (LOCF) was greater in the azilsartan group than in the candesartan group in all other subgroups (see Supplementary Information).
Safety/tolerability
The study drugs were equally well tolerated and there were no clear differences in the incidences of treatment-emergent adverse events (TEAEs) between the two treatment groups. TEAEs were reported by 58.1% of patients (182/313) who received azilsartan and 52.4% (162/309) who received candesartan (Table 3). The vast majority of TEAEs were either mild or moderate in intensity in the two groups (181 of 182 in the azilsartan group; 160 of 162 in the candesartan group). No clear trend of time- or dose-dependency in the incidence of TEAEs was evident in either treatment group (data not shown). No deaths occurred during the study. Discontinuations due to adverse events and serious adverse events were infrequent in both groups.
The most common TEAEs occurring in 3% or more of the patients in the azilsartan group were nasopharyngitis (18.8% in the azilsartan group vs. 16.2% in the candesartan group), upper respiratory tract inflammation (5.1% vs. 4.2%, respectively), and pharyngitis (3.2% vs. 2.3%, respectively). The overall incidence of hypotension-related events (dizziness, dizziness postural, syncope, vertigo and vertigo positional) was comparable to the two drugs— 11 of 313 patients (3.5%) who received azilsartan as compared with 10 of 309 patients (3.2%) who received candesartan. Syncope was reported in one patient in the candesartan group.
Adverse events considered treatment-related were infrequent in both groups, but were slightly more common with azilsartan than with candesartan (7.3% vs. 1.9%; Table 3). This was mainly due to slightly higher incidences of postural dizziness (1.6% vs. 0.3%) and increased blood uric acid levels (1.6% vs. 0%), although they were infrequent and were not of clinical concern as they did not lead to syncope or gout.
There were no remarkable findings of clinical concern in laboratory test results, vital signs, body weight and 12-lead electrocardiogram findings.
Discussion
The findings of the study showed that azilsartan (20–40 mg once daily) provided a significantly greater reduction from baseline of clinic-measured BP (both sitting trough SBP and sitting trough DBP) than candesartan (8–12 mg once daily) in Japanese patients with grade I–II essential hypertension at all time points from weeks 2 to 16 over the treatment period. In addition, the proportions of clinical responders and patients who were categorized as well-controlled at weeks 8 and 16 were significantly higher in the azilsartan group than in the candesartan group. When the time-courses of BP changes with two ARBs were evaluated by ABPM at 14 weeks, azilsartan was also found to provide a significantly greater reduction from baseline in mean SBP and DBP than candesartan during the 24-h time period, as well as in the daytime during waking hours, at night-time during sleep, and in the early morning (SBP), indicating a more sustained duration of action. Moreover, trough-to-peak ratios for both DBP and SBP on ABPM at week 14 of treatment were higher in the azilsartan group than in the candesartan group, providing further evidence of a longer duration of action of azilsartan relative to candesartan.19
The longer duration of antihypertensive efficacy of azilsartan was not at the expense of diminished tolerability, as the two ARBs were equally well tolerated in this study. The majority of TEAEs were mild in severity, and the most commonly reported events with both drugs were nasopharyngitis, upper respiratory tract inflammation and pharyngitis. There was a slightly higher incidence of treatment-related AEs with azilsartan than with candesartan (7.3% vs. 1.9%), mainly as a result of slightly higher incidences of postural dizziness (1.6% vs. 0.3%) and increased blood uric acid levels (1.6% vs. 0%). However, these events were generally of mild intensity and resolved without intervention and, importantly, were not of clinical concern as they did not lead to syncope or gout. Overall, treatment-related AEs were infrequent in the two groups. There was no clear trend of time- or dose-dependency in the incidence of TEAEs with either treatment, and there were no remarkable findings of clinical concern in laboratory test results, vital signs, body weight and 12-lead electrocardiogram findings.
Earlier clinical trials conducted outside of Japan that compared azilsartan medoxomil (a prodrug of azilsartan that is rapidly converted to the active form, azilsartan, during the absorption phase) with other ARBs, have reported similar findings to our study. In a multicenter, randomized, double-blind study that compared azilsartan medoxomil and olmesartan medoxomil in US patients with primary hypertension, azilsartan medoxomil was significantly more effective in lowering mean 24-h SBP than olmesartan medoxomil.20 Similarly, in a multicenter, randomized, double-blind study in patients with grade I–II hypertension conducted in Latin American countries and the USA, treatment with azilsartan medoxomil was significantly more effective than valsartan and olmesartan in lowering mean 24-h SBP.21 In both trials, the safety profile of azilsartan medoxomil was similar to that of the ARBs and the placebo with which it was compared.
The principal limitations of the present study were that only patients with grade I or II essential hypertension (without cardiovascular disease or significant renal impairment) were eligible for enrollment and the relatively short treatment duration, which preclude its extrapolation to other categories of hypertensive patients and any definitive conclusions regarding its target organ-protective effects. However, with regard to target organ protection, the Japanese People’s Health Promotion Campaign for the 21st Century (Healthy Japan 21)22 has estimated that a 2 mm Hg decrease in the national SBP level would lead to a 6.4% decrease in the prevalence of stroke and a 5.4% decrease in that of ischemic heart disease (IHD), as well as a decrease of approximately 9000 in the number of deaths from stroke and approximately 4000 in the number of deaths from IHD. Therefore, the LS mean difference in SBP recorded in this study of −4.4 mm Hg between the treatment groups after 16 weeks of treatment is likely to be clinically relevant and would be expected to contribute to a decrease in the morbidity and mortality rates related to cerebrovascular disease and IHD. Moreover, as the average BP level over 24 h, nocturnal and early morning BP are reported to be more closely associated with hypertensive target organ damage and cardiovascular events than clinic-measured BP,4, 5, 6 the finding that azilsartan provided a significantly greater reduction from baseline to week 14 in mean SBP and DBP than candesartan during a 24-h time period, as well as in the daytime during waking hours, night-time hours during sleep, and in the early morning (SBP) suggests that it provides greater protection against cardiovascular events.
In conclusion, this study has shown that once-daily administration of azilsartan produces a 24-h sustained antihypertensive effect that is more potent than that of candesartan in Japanese patients with grade I–II essential hypertension but at an equivalent level of safety. Consequently, azilsartan administered once daily could provide higher rates of hypertension control during a 24-h period (including the night and early morning hours) and might be expected to give greater protection against cardiovascular events in patients with essential hypertension, although this remains to be proven in prospectively designed clinical studies.
References
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R . Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360: 1903–1913.
Japanese Society of Hypertension Committee,. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2009). Hypertens Res 2009; 32: 3–107.
Ohkubo T, Obara T, Funahashi J, Kikuya M, Asayama K, Metoki H, Oikawa T, Takahashi H, Hashimoto J, Totsune K, Imai Y, J-HOME Study Group. Control of blood pressure as measured at home and office, and comparison with physicians’ assessment of control among treated hypertensive patients in Japan: First Report of the Japan Home versus Office Blood Pressure Measurement Evaluation (J-HOME) study. Hypertens Res 2004; 27: 755–763.
Staessen JA, Thijs L, Fagard R, O’Brien ET, Clement D, de Leeuw PW, Mancia G, Nachev C, Palatini P, Parati G, Tuomilehto J, Webster J . Predicting cardiovascular risk using conventional vs ambulatory blood pressure in older patients with systolic hypertension. Systolic Hypertension in Europe Trial Investigators. JAMA 1999; 282: 539–546.
Kikuya M, Ohkubo T, Asayama K, Metoki H, Obara T, Saito S, Hashimoto J, Totsune K, Hoshi H, Satoh H, Imai Y . Ambulatory blood pressure and 10-year risk of cardiovascular and noncardiovascular mortality: the Ohasama study. Hypertension 2005; 45: 240–245.
Asayama K, Ohkubo T, Kikuya M, Obara T, Metoki H, Inoue R, Hara A, Hirose T, Hoshi H, Hashimoto J, Totsune K, Satoh H, Imai Y . Prediction of stroke by home ‘morning’ versus ‘evening’ blood pressure values: the Ohasama study. Hypertension 2006; 48: 737–743.
Obara T, Ito K, Ohkubo T, Shibamiya T, Shinki T, Nakashita M, Hara A, Metoki H, Inoue R, Asayama K, Kikuya M, Mano N, Imai Y, J-HOME Study Group. Uncontrolled hypertension based on morning and evening home blood pressure measurements from the J-HOME study. Hypertens Res 2009; 32: 1072–1078.
Ishikawa J, Kario K, Eguchi K, Morinari M, Hoshide S, Ishikawa S, Shimada K, J-MORE Group. Regular alcohol drinking is a determinant of masked morning hypertension detected by home blood pressure monitoring in medicated hypertensive patients with well-controlled clinic blood pressure: the Jichi Morning Hypertension Research (J-MORE) study. Hypertens Res 2006; 29: 679–686.
Smith DH . Comparison of angiotensin II type 1 receptor antagonists in the treatment of essential hypertension. Drugs 2008; 68: 1207–1225.
Ogihara T, Nakao K, Fukui T, Fukiyama K, Ueshima K, Oba K, Sato T, Saruta T, Candesartan Antihypertensive Survival Evaluation in Japan Trial Group. Effects of candesartan compared with amlodipine in hypertensive patients with high cardiovascular risks: candesartan antihypertensive survival evaluation in Japan trial. Hypertension 2008; 51: 393–398.
Weber MA, Julius S, Kjeldsen SE, Brunner HR, Ekman S, Hansson L, Hua T, Laragh JH, McInnes GT, Mitchell L, Plat F, Schork MA, Smith B, Zanchetti A . Blood pressure dependent and independent effects of antihypertensive treatment on clinical events in the VALUE Trial. Lancet 2004; 363: 2049–2051.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint Reduction in Hypertension Study (LIFE): a randomised trial against atenolol. Lancet 2002; 359: 995–1003.
Demers C, McMurray JJ, Swedberg K, Pfeffer MA, Granger CB, Olofsson B, McKelvie RS, Ostergren J, Michelson EL, Johansson PA, Wang D, Yusuf S, CHARM Investigators. Impact of candesartan on nonfatal myocardial infarction and cardiovascular death in patients with heart failure. JAMA 2005; 294: 1794–1798.
McMurray JJ, Ostergren J, Swedberg K, Granger CB, Held P, Michelson EL, Olofsson B, Yusuf S, Pfeffer MA, CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362: 767–771.
Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861–869.
Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851–860.
Ojima M, Igata H, Tanaka M, Sakamoto H, Kuroita T, Kohara Y, Kubo K, Fuse H, Imura Y, Kusumoto K, Nagaya H . In vitro antagonistic properties of a new angiotensin type 1 receptor blocker, azilsartan, in receptor binding and function studies. J Pharmacol Exp Ther 2011; 336: 801–808.
Abe K, Kuwashima I, Imai J, Shimada K, Shimamoto K, Fujita T, Saito I, Hisashiro T, Fukiyama K . An effect of long-acting calcium channel blocker, barnidipine on 24 h blood pressure: A final result of Japanese multicenter study on barnidipine with ABPM (J-MUBA). Ther Res 1999; 20: 2697–2713.
Meredith PA . Importance of trough: peak ratios in treatment of blood pressure in post-myocardial infarction and heart failure settings. J Hum Hypertens 1997; 11: 39–43.
Bakris GL, Sica D, Weber M, White WB, Roberts A, Perez A, Cao C, Kupfer S . The comparative effects of azilsartan medoxomil and olmesartan on ambulatory and clinic blood pressure. J Clin Hypertens 2011; 13: 81–88.
White WB, Weber MA, Sica D, Bakris GL, Perez A, Cao C, Kupfer S . Effects of the angiotensin receptor blocker azilsartan medoxomil versus olmesartan and valsartan on ambulatory and clinic blood pressure in patients with stages 1 and 2 hypertension. Hypertension 2011; 57: 413–420.
Japan Health Promotion and Fitness Foundation. [Report of People's Health Promotion Campaign for the 21st Century (Healthy Japan 21)]. 2000: 8-22 [Japanese].
Acknowledgements
This study was supported by Takeda Pharmaceutical Company Limited (Osaka, Japan). We acknowledge the investigators, other staff members and patients who made this study possible. We also appreciate the support provided by Mr Jouichi Ichise and Mr Masataka Igeta Takeda Pharmaceutical Company Limited, and ContentEdNet for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
HR served as the medical expert for this study, and received honoraria from Takeda Pharmaceutical Company for lectures he gave during the study period. KE, KS and YI are all employees of Takeda Pharmaceutical Company. HR has no conflicts of interest regarding the content of this article other than as stated above.
Additional information
Supplementary Information accompanies the paper on Hypertension Research website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rakugi, H., Enya, K., Sugiura, K. et al. Comparison of the efficacy and safety of azilsartan with that of candesartan cilexetil in Japanese patients with grade I–II essential hypertension: a randomized, double-blind clinical study. Hypertens Res 35, 552–558 (2012). https://doi.org/10.1038/hr.2012.8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.8
Keywords
This article is cited by
-
Safety and efficacy of azilsartan in paediatric patients with hypertension: a phase 3, single-arm, open-label, prospective study
Clinical and Experimental Nephrology (2022)
-
Catheter-based ultrasound renal denervation in patients with resistant hypertension: the randomized, controlled REQUIRE trial
Hypertension Research (2022)
-
Antihypertensive effect of azilsartan versus olmesartan in patients with essential hypertension: a meta-analysis
Irish Journal of Medical Science (1971 -) (2019)
-
Pharmacokinetics of a Single Dose of Azilsartan in Pediatric Patients: A Phase 3, Open-Label, Multicenter Study
Advances in Therapy (2018)
-
Azilsartan: from bench to bedside
Drugs & Therapy Perspectives (2016)
