
Abstract
High serum uric acid level (SUA) and chronic kidney disease (CKD) are risk factors for cardiovascular events (CVEs). However, their interactions as cardiovascular risk factors remain unknown. This subanalysis of the Japan Hypertension Evaluation with Angiotensin II Antagonist Losartan Therapy (J-HEALTH) study included 7629 patients, in whom the serum creatinine level was measured at least twice. The study examined the impact of hyperuricemia (SUA ⩾7 mg dl−1) on CVE according to the level of renal dysfunction and whether early changes in SUA predicted future glomerular filtration rates (GFRs). The mean follow-up period was 3.1 years. The patients were divided into three groups according to the baseline estimated GFR (eGFR): groups A, B and C with eGFR <45, 45–59 and ⩾60 ml min−1 per 1.73 m2, respectively. eGFR increased from 38.1 to 57.6, from 52.8 to 67.5 and from 74.7 to 80.7 ml min−1 per 1.73 m2 in groups A, B and C, respectively. In non-hyperuricemic patients, the CVE rate was 10.83, 4.98 and 4.21/1000 person-years in groups A, B and C, respectively, while in hyperuricemic patients, the corresponding values were 14.18, 17.02 and 5.93. Thus, hyperuricemia increased the risk of CVE only in group B (relative risk (RR) 3.43 (95% confidence interval (CI) 1.55 to 7.60); P<0.002). The final change in the eGFR was negatively correlated with the change in SUA from baseline to year 1 (P<0.001). CVEs were more frequent in those with a decrease in eGFR. Hyperuricemia may be a major determinant of increased cardiovascular risk in CKD stage 3A, and SUA may be involved in the progression of CKD. Changes in the GFR influence the rate of CVE.
Similar content being viewed by others

Introduction
Chronic kidney disease (CKD) is associated with an increase in cardiovascular morbidity and mortality.1 A reduced glomerular filtration rate (GFR) and albuminuria are independently associated with worse cardiovascular and renal outcomes in various populations.2, 3 In the recently modified classification of CKD published in Kidney Disease: Improving Global Outcomes (KDIGO), stage 3 of CKD was subdivided into two stages (stage 3A and 3B with GFR 45–59 and 30–44 ml min−1 per 1.73 m2, respectively).4 It was shown that cardiovascular risk increases sharply when the estimated GFR (eGFR) falls below 45 ml min−1 per 1.73 m2 (ref. 5). Moreover, the new classification of CKD emphasizes that underlying diseases play an important role in cardio-renal outcomes in CKD.
Many studies suggest that an increased serum uric acid level (SUA) is associated with cardiovascular events (CVEs).6, 7, 8 Hyperuricemia is highly prevalent in patients with CKD. Thus, SUA may have a role in the increased cardiovascular mortality and morbidity associated with CKD. However, scant data exist on whether the impact of hyperuricemia on CVE differs according to the degree of renal dysfunction.
The issue of whether hyperuricemia is an independent risk factor for CKD remains controversial. A large number of cross-sectional studies have shown that hyperuricemia is independently and strongly associated with the prevalence of CKD.9, 10, 11 While prospective longitudinal studies also suggest that hyperuricemia may be an independent risk factor for CKD, its impact varies substantially among reports.12, 13, 14, 15 In addition, there is little information regarding whether SUA influences future renal function in CKD patients.
The Japan Hypertension Evaluation with Angiotensin II Antagonist Losartan Therapy (J-HEALTH) study was a nationwide, prospective, multicenter, observational study that examined the safety and effectiveness of losartan-based antihypertensive therapy in 31 515 previously untreated hypertensive patients.16 All the patients were treated with losartan, which has been shown to lower SUA. In this subanalysis, we used data from 7629 patients whose SUA was measured at least twice and investigated whether the impact of SUA on CVE differs among three KDIGO categories of eGFR (<45, 45–59, ⩾60 ml min−1 per 1.73 m2), whether early changes in SUA during losartan treatment predict later GFR and whether shifts in GFR are associated with CVE.
Methods
Study design and setting
The details of the J-HEALTH Study have been previously described.16 A total of 31 515 patients were screened between June 2000 and December 2001 in all of Japan’s 47 prefectures at 3755 institutions by 4149 physicians. Among the study participants, 7629 patients in whom the serum creatinine level was measured at least twice were eligible for analysis.
Eligible patients were men or women >20 years of age with hypertension and who had not received any antihypertensive agents within the previous month. Before enrollment, each patient was informed of the purpose and methods of the study as well as the effects and possible risks of therapy, their right to withdraw from the study at any time and the measures taken to protect privacy. Each patient provided verbal informed consent. This trial was registered at https://center.umin.ac.jp as UMIN000006149.
The patients were initially treated with 25–50 mg of losartan once daily. If necessary, the dose could be increased up to 100 mg once daily. The addition of other antihypertensive agents was allowed 3 months after the start of losartan therapy. No restrictions were placed on the treatment of complications. Patients were followed for a maximum of 5 years, with a mean follow-up of 3.1 years.
Blood Pressure (BP) was measured by the methods typically used at each institution. After starting losartan therapy, BP was measured every 3 months during treatment. Standard laboratory tests were performed every 6 months. These were carried out in accordance with the routine methods used at each institution.
Data sources/measurement
The eGFR was calculated using the following equation for Japanese:17

CKD was defined as present when the eGFR was <60 ml min−1 per 1.73 m2. Urinary protein was examined by a dipstick method and classified as negative (−), ±, 1+ or 2+. The primary end points were the occurrence of CVE (stroke, myocardial infarction or sudden death) and changes in the eGFR.
Statistical methods
Variables were compared using the t-test, the χ2-test, or an analysis of variance. The results were expressed as the mean±s.d., and differences were considered statistically significant at P<0.05. An assessment of the overall results was based on a survival analysis. The eGFR and BP data obtained after the onset of any disease (including a primary end point) were excluded from the analysis. Subgroups were divided based on eGFR values measured before and at the end of treatment. We used the Cox proportional hazards model to assess the relation between CVE and eGFR values. A multiple linear regression analysis was used to explore determinants of changes in eGFR; predictors of such changes (baseline eGFR, diabetes mellitus and stroke) were included in the original model. We used SAS software package Version 8.02 (SAS Institute, Cary, NC, USA) to perform all statistical analyses.
Results
Baseline characteristics
The patients were divided into three groups: A, B and C, with eGFR <45, 45–59 and ⩾60 ml min−1 per 1.73 m2, respectively. The baseline characteristics of the subjects are shown in Table 1. Approximately 11, 38 and 51% of the patients belonged to groups A, B and C, respectively. The majority of the CKD patients were in stage 3 (n=3646); only a few were in stage 4 (n=75) or 5 (n=12). The CKD patients were older and had a higher prevalence of cerebrovascular and cardiovascular complications. Systolic blood pressure (SBP) and the prevalence of diabetes and dyslipidemia were similar among the groups. The rates of smoking and regular alcohol intake were lower in patients with CKD. Major concomitant drugs were similar in all groups, except for those in group A (the lowest eGFR); the patients in this group took more diuretics and uric acid–lowering drugs compared with those in the other two groups. Antihypertensive and lipid-lowering drugs were the most frequently used concomitant drugs in all patients, and calcium antagonists were prescribed for 9.2% of patients.
Blood pressure and urinary protein excretion
The mean SBP/diastolic BP decreased similarly in the three groups, and BP was well-controlled throughout the study period. In group A, the SBP/diastolic BP decreased from 165.6±17.3/90.2±11.8 mm Hg at baseline to 145.7±16.5/80.8±10.8 mm Hg after 3 months and to 139.9±16.9/79.8±10.9 mm Hg after 60 months of treatment. In group B, the SBP/diastolic BP decreased from 165.3±16.5/93.2±11.1 mm Hg to 145.6±15.5/83.1±10.3 mm Hg after 3 months and to 135.3±11.9/76.7±8.4 mm Hg after 60 months. In group C, the SBP/diastolic BP decreased from 165.3±16.4/94.9±11.3 mm Hg to 144.8±15.6/84.1±10.4 mm Hg after 3 months and to 135.4±10.8/79.5±8.0 mm Hg after 60 months.
The percentage of patients with positive urinary protein (± or above with dipstick test) decreased from 19.7% at baseline to 7.9% at the end of treatment. Improvement was apparent at 6 months, and continued throughout the treatment period.
Changes in eGFR
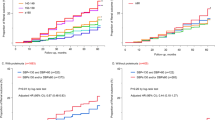
Changes in eGFR are shown in Figure 1. In group C, the eGFR slightly decreased initially from a baseline value of 74.7±15.8 ml min−1 per 1.73 m2 (by 2.7 ml−1 min per 1.73 m2 at 1 year) and then gradually increased to a final value of 80.7±23.3 mlmin−1 per 1.73 m2 at the end of the study. In group B, eGFR increased steadily from 52.8±4.1 to 67.5±15.7 ml min−1 per 1.73 m2, while it increased from 38.1±6.7 to 57.6±18.1 ml min−1 per 1.73 m2 in group A.

Serial changes in the estimated glomerular filtration rate (eGFR) in hypertensive patients with or without chronic kidney disease.
Relationship between eGFR and CVE
A total of 141 CVEs occurred, including 103 strokes and 38 myocardial infarcts. We analyzed the incidence of CVEs according to the eGFR at baseline as well as at the end of treatment (Table 2). The event rate became higher as the baseline eGFR declined. The relative risk (RR) of CVE in group A patients was 2.81 (95% confidence interval (CI), 1.80–4.39; P<0.001) compared with that of group C patients. The final eGFR values had even stronger influences on the rate of CVE than the baseline eGFR value did. Compared with the event rate in patients with a final eGFR ⩾60 ml min−1 per 1.73 m2, the RRs for CVE in the patients with a final eGFR <45 and 45–59 ml min−1 per 1.73 m2 were 3.24 (95% CI 2.06–5.08; P<0.001) and 2.25 (95% CI 1.54–3.28; P<0.001), respectively.
We also analyzed the incidence of CVE according to changes in the eGFR. Decreases in the eGFR were associated with higher event rates as compared with those observed in patients with an increase or no change in the eGFR. This was particularly evident in group A patients with a baseline eGFR <45 ml min−1 per 1.73 m2.
Serum uric acid and CVE
In group A, SUA decreased significantly from 5.81±1.56 mg dl−1 at baseline to 5.53±1.50 mg dl−1 at 6 months and remained at the same level thereafter. In groups B and C, SUA dropped from a baseline value of 5.31±1.53 and 5.21±1.51 mg dl−1 to 5.09±1.40 and 5.03±1.37 mg dl−1 after 6 months of treatment, respectively.
Table 3 shows the demographics of the patients according to the change in SUA from baseline to 1 year of losartan treatment. Of note, the baseline SUA was higher in patients whose SUA decreased more than 0.5 mg dl−1 compared with patients whose SUA remained unchanged or increased. In addition, there were large differences in final change in the eGFR from baseline to the end of follow-up, and the increase in the eGFR was greatest in the group with SUA decrement greater than 0.5 mg dl−1.
The impact of hyperuricemia (SUA ⩾7 mg dl−1) on CVE was first examined in the overall patient population. The CVE rate was found to be 2.06 (95% CI 1.21–3.52; P=0.008) times higher in the patients with hyperuricemia than in those without hyperuricemia. Figure 2 shows the CVE rate stratified by the eGFR in the presence and absence of hyperuricemia. In the absence of hyperuricemia (SUA <7 mg dl−1), the CVE rate was 10.83, 4.98 and 4.21/1000 person-years in groups A, B and C, respectively; the event rate was significantly (P=0.003) higher in group A as compared with groups B and C, while it was similar in groups B and C. In the presence of hyperuricemia, however, the event rate in group B (17.02) was as high as that of group A (14.18), and it was significantly (P<0.01) higher than that of group C (5.93). Thus, hyperuricemia increased the risk of CVE only in group B (RR 3.43 (95% CI 1.55–7.60); P=0.002).

Impact of hyperuricemia on cardiovascular event rate. eGFR, estimated glomerular filtration rate.
To examine possible influences of uric acid–lowering drugs and/or diuretics on the relationship between hyperuricemia and CVE, we performed the same analyses excluding those patients taking any of these drugs. The results were essentially the same, in which hyperuricemia increased the risk of CVE only in group B (RR 2.81 (95% CI 1.07–7.33); P=0.036), but not in group A (RR 1.36 (95% CI 0.46–4.01); P=0.58) or group C (RR 1.58 (95% CI 0.56–4.47); P=0.392).
Parameters associated with changes in GFR
To explore the factors that may contribute to the observed improvement in eGFR, we performed a multiple linear regression analysis for changes in the eGFR. As shown in Table 4, the SBP during treatment, the SUA during treatment, the change (Δ) in SUA at 1 year and in urinary protein at 1 year were all significantly associated with a ΔeGFR from baseline to the end of the study in all three groups. ΔSUA and proteinuria stage at 1 year were predictive factors when the model included SBP throughout the study period, ΔSUA and proteinuria stage at 1 year. Therefore, ΔSUA and proteinuria stage were confirmed to be independent predictors of ΔeGFR from baseline to the end of the study.
The correlations between ΔSUA at 1 year and ΔeGFR from baseline to the end of the study, in both CKD and non-CKD patients, were strong and negative. Each 1-mg decrease in SUA from baseline to 1 year was associated with final increases in the eGFR of 2.71 ml min−1 per 1.73 m2 for the patients with a baseline eGFR <45 ml min−1 per 1.73 m2, 1.84 ml min−1 per 1.73 m2 for those with a baseline eGFR of 45–59 ml min−1 per 1.73 m2 and 1.31 ml min−1 per 1.73 m2 for those with a baseline eGFR ⩾60 ml min−1 per 1.73 m2.
Discussion
In the J-HEALTH study, hypertensive subjects were treated with losartan, an angiotensin-receptor blocker that has a unique uricosuric action.18, 19 This subanalysis confirms that the baseline eGFR, as well as changes in the eGFR, have marked influences on cardiovascular mortality and morbidity. Hyperuricemia seems to enhance cardiovascular risks strongly in patients with an eGFR of 45–59 ml min−1 per 1.73 m2 (CKD stage 3A). An early reduction in SUA occurred before a substantial change in the eGFR, and this decline was closely associated with a future increase in the eGFR. Thus, SUA may have a role in the development of renal dysfunction in hypertensive patients.
In the recently modified classification of CKD by KDIGO, CKD stage 3 was subdivided into stages 3A and 3B, taking into consideration the epidemiological observations that cardiovascular risk increases sharply when the eGFR falls below 45 ml min−1 per 1.73 m2. Consistently, we also observed greatly increased CVE in patients with a baseline eGFR <45 ml min−1 per 1.73 m2. In addition, we observed that the RR of CVE increased in patients who had a decrease in eGFR, and this was particularly evident in those with a baseline eGFR <45 ml min−1 per 1.73 m2. These results indicate that not only the baseline eGFR but also the change in eGFR is an important determinant of CVE. Consistent with our study, Khan et al.20 reported that the rate of decline in the eGFR paralleled mortality in patients with left ventricular systolic dysfunction.
Few studies have examined the interaction of renal function and SUA in terms of cardiovascular risk, and the results of these studies are contradictory. Neri et al.21 reported that SUA was a strong independent correlate of CVE in CKD patients but not in non-CKD patients, whereas Navaneethan et al.22 reported that the presence of CKD attenuated the association of SUA with mortality. In this study, we examined the impact of hyperuricemia on CVE in three groups with an eGFR <45, 45–59 and ⩾60 ml min−1 per 1.73 m2, because studies have established that there are clear differences in the CVE rate between CKD stages 3A and 3B. We observed that in the absence of hyperuricemia, the CVE rate was almost identical in those without CKD and in those with CKD stage 3A (Figure 2), suggesting that CKD stage 3A alone, without hyperuricemia, may not be a strong risk factor for CVE. However, once the patients with CKD stage 3A had hyperuricemia, the risk of CVE was dramatically elevated. This may suggest that hyperuricemia may account for much of the cardiovascular risk associated with CKD stage 3A. On the other hand, hyperuricemia had no impact on CVE in those patients with an eGFR ⩾60 or <45 ml min−1 per 1.73 m2. It may be that in patients with a very high risk of CVE, such as those with an eGFR <45 ml min−1 per 1.73 m2, hyperuricemia may have few additional effects on enhancing CVE. Alternatively, in those with very low risk of CVE, such as those with an eGFR ⩾60 ml min−1 per 1.73 m2, there may be few or no other factors with which hyperuricemia acts synergistically to enhance CVE risks. We recently found that the plasma methylglyoxal level is elevated in CKD and that it may be involved in vascular injury in animals and humans.23, 24, 25 Interestingly, uric acid has an amine residue that can react with the carbonyl residue in compounds such as methylglyoxal. In addition, oxidative stress and inflammation are enhanced even in the early stages of CKD.26 Thus, uric acid may exert negative effects in the stage 3A CKD patients by interacting synergistically with oxidative stress, inflammatory molecules and methylglyoxal.
Antihypertensive treatment with inhibitors of the renin–angiotensin system has been shown to reduce proteinuria and retard the rate of decline of renal function in hypertensive CKD patients.27, 28 In this study, we have shown that losartan-based antihypertensive treatment can increase the eGFR in hypertensive patients. Further analyses of the factors associated with changes in the eGFR identified changes in SUA as a novel risk factor, in addition to the well-known risk factors for the decline of renal function.29, 30 It is of note that a change in SUA was as powerful a risk factor as proteinuria status, which has been shown to be the strongest risk factor for end-stage renal failure. An early reduction of SUA from baseline to 1 year was strongly correlated with a future increase in the eGFR from baseline to the end of the study. Because the decreases in SUA occurred with few changes in eGFR, it may not be secondary to an increased eGFR. It may be assumed that decreases in SUA induced by losartan improved renal function in the present study. Indeed, studies have reported that allopurinol reduced SUA and retarded the progression of renal dysfunction.31, 32, 33 The importance of the reduction of SUA in long-term renal risk reduction was also confirmed in a post-hoc analysis of 1342 patients with type 2 diabetes mellitus and nephropathy in the Reduction of Endpoints in Non-Insulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study.34 Taken together with these studies, our study provides strong support for the role of SUA in kidney damage in hypertensive patients.
The mechanism by which a decrease in SUA results in improved renal function is not clear from the present study. Experimental studies have shown that hyperuricemia causes endothelial, arteriolar and tubular interstitial damage in the kidney by increasing oxidative stress and inflammation.35, 36 Price et al.37 reported that uric acid is transported into endothelial cells via urate reabsorptive transporter-1, and it then induces oxidative stress. In addition, it has been reported that hyperuricemia increases juxtaglomerular renin expression and decreases macula densa neuronal nitric oxide synthase expression.38 Thus, uric acid may cause renal injury by interacting synergistically with the renin–angiotensin system, oxidative stress and inflammatory molecules.
Our study has several limitations. The first is its observational design, which precludes the assignment of causation. Although BP levels were well-matched among the three groups, the medications used in addition to the initial 25–50 mg dose of losartan were prescribed at the discretion of participating physicians. We did not set a specific target BP or rules for additional drugs nor were we able to centralize the measurement of serum creatinine. Various guidelines for hypertension treatment recommend a target BP for CKD patients of <130/80 mm Hg. However, we could not achieve this goal. In addition, the CVE rate was low, particularly among non-CKD patients, which may have obscured the potential effects of hyperuricemia on CVEs.
In conclusion, the J-HEALTH study has demonstrated that losartan-based antihypertensive treatment increases the eGFR and reduces BP, proteinuria and SUA. The early reduction of proteinuria and SUA may predict future improvement of renal function, and the improvement of renal function greatly affects the occurrence of CVE, particularly in CKD patients. Hyperuricemia seems to be a major contributor to increased cardiovascular risks in early-stage CKD. These findings may be relevant to evaluating and managing cardiorenal risks in CKD patients. Further investigations are needed to clarify the role of uric acid in the pathophysiology of CKD.
References
Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, Parfrey P, Pfeffer M, Raij L, Spinosa DJ, Wilson PW . American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 2003; 42: 1050–1065.
Chronic Kidney Disease Prognosis Consortium; Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Gansevoort RT . Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010; 375: 2073–2081.
Hemmelgarn BR, Manns BJ, Lloyd A, James MT, Klarenbach S, Quinn RR, Wiebe N, Tonelli M . Alberta Kidney Disease Network. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 2010; 303: 423–429.
Levey AS, de Jong PE, Coresh J, El Nahas M, Astor BC, Matsushita K, Gansevoort RT, Kasiske BL, Eckardt KU . The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011; 80: 17–28.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY . Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305.
Fang J, Alderman MH . Serum uric acid and cardiovascular mortality the NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey. JAMA 2000; 283: 2404–2410.
Niskanen LK, Laaksonen DE, Nyyssönen K, Alfthan G, Lakka HM, Lakka TA, Salonen JT . Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study. Arch Intern Med 2004; 164: 1546–1551.
Liese AD, Hense HW, Löwel H, Döring A, Tietze M, Keil U . Association of serum uric acid with all-cause and cardiovascular disease mortality and incident myocardial infarction in the MONICA Augsburg cohort. World Health Organization Monitoring Trends and Determinants in Cardiovascular Diseases. Epidemiology 1999; 10: 391–397.
Chen YC, Su CT, Wang ST, Lee HD, Lin SY . A preliminary investigation of the association between serum uric acid and impaired renal function. Chang Gung Med J 2009; 32: 66–71.
Chonchol M, Shlipak MG, Katz R, Sarnak MJ, Newman AB, Siscovick DS, Kestenbaum B, Carney JK, Fried LF . Relationship of uric acid with progression of kidney disease. Am J Kidney Dis 2007; 50: 239–247.
See LC, Kuo CF, Chuang FH, Li HY, Chen YM, Chen HW, Yu KH . Serum uric acid is independently associated with metabolic syndrome in subjects with and without a low estimated glomerular filtration rate. J Rheumatol 2009; 36: 1691–1698.
Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S . Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis 2004; 44: 642–650.
Domrongkitchaiporn S, Sritara P, Kitiyakara C, Stitchantrakul W, Krittaphol V, Lolekha P, Cheepudomwit S, Yipintsoi T . Risk factors for development of decreased kidney function in a southeast Asian population: a 12-year cohort study. J Am Soc Nephrol 2005; 16: 791–799.
Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R . Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol 2008; 19: 2407–2413.
Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS . Uric acid and incident kidney disease in the community. J Am Soc Nephrol 2008; 19: 1204–1211.
Naritomi H, Fujita T, Ito S, Ogihara T, Shimada K, Shimamoto K, Tanaka H, Yoshiike N . Design and baseline characteristics of an observational study in Japanese patients with hypertension: Japan Hypertension Evaluation with Angiotensin II Antagonist Losartan Therapy (J-HEALTH). Hypertens Res 2007; 30: 807–814.
Matsuo S, Imai E, Horio M, Yasuda Y, Tomita K, Nitta K, Yamagata K, Tomino Y, Yokoyama H, Hishida A . Collaborators developing the Japanese equation for estimated GFR. Revised equations for estimated GFR from serum creatinine in Japan. Am J Kidney Dis 2009; 53: 982–992.
Hamada T, Ichida K, Hosoyamada M, Mizuta E, Yanagihara K, Sonoyama K, Sugihara S, Igawa O, Hosoya T, Ohtahara A, Shigamasa C, Yamamoto Y, Ninomiya H, Hisatome I . Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens 2008; 21: 1157–1162.
Hatch M, Freel RW, Shahinfar S, Vaziri ND . Effects of the specific angiotensin II receptor antagonist losartan on urate homeostasis and intestinal urate transport. J Pharmacol Exp Ther 1996; 276: 187–193.
Khan NA, Ma I, Thompson CR, Humphries K, Salem DN, Sarnak MJ, Levin A . Kidney function and mortality among patients with left ventricular systolic dysfunction. J Am Soc Nephrol 2006; 17: 244–253.
Neri L, Rocca Rey LA, Lentine KL, Hinyard LJ, Pinsky B, Xiao H, Dukes J, Schnitzler MA . Joint association of hyperuricemia and reduced GFR on cardiovascular morbidity: a historical cohort study based on laboratory and claims data from a national insurance provider. Am J Kidney Dis 2011; 58: 398–408.
Navaneethan SD, Beddhu S . Associations of serum uric acid with cardiovascular events and mortality in moderate chronic kidney disease. Nephrol Dial Transplant 2009; 24: 1260–1266.
Nakayama K, Nakayama M, Iwabuchi M, Terawaki H, Sato T, Kohno M, Ito S . Plasma alpha-oxoaldehyde levels in diabetic and nondiabetic chronic kidney disease patients. Am J Nephrol 2008; 28: 871–878.
Guo Q, Mori T, Jiang Y, Hu C, Osaki Y, Yoneki Y, Sun Y, Hosoya T, Kawamata A, Ogawa S, Nakayama M, Miyata T, Ito S . Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague-Dawley rats. J Hypertens 2009; 27: 1664–1671.
Ogawa S, Nakayama K, Nakayama M, Mori T, Matsushima M, Okamura M, Senda M, Nako K, Miyata T, Ito S . Methylglyoxal is a predictor in type 2 diabetic patients of intima-media thickening and elevation of blood pressure. Hypertension 2010; 56: 471–476.
Libetta C, Sepe V, Esposito P, Galli F, Dal Canton A . Oxidative stress and inflammation: implications in uremia and hemodialysis. Clin Biochem 2011; 44: 1189–1198.
Wright JT, Bakris G, Greene T, Agodoa LY, Appel LJ, Charleston J, Cheek D, Douglas-Baltimore JG, Gassman J, Glassock R, Hebert L, Jamerson K, Lewis J, Phillips RA, Toto RD, Middleton JP, Rostand SG . African American Study of Kidney Disease and Hypertension Study Group. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA 2002; 288: 2421–2431.
Jafar TH, Stark PC, Schmid CH, Landa M, Maschio G, de Jong PE, de Zeeuw D, Shahinfar S, Toto R, Levey AS . AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003; 139: 244–252.
Bakris GL, Williams M, Dworkin L, Elliott WJ, Epstein M, Toto R, Tuttle K, Douglas J, Hsueh W, Sowers J . Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36: 646–661.
van der Velde M, Halbesma N, de Charro FT, Bakker SJ, de Zeeuw D, de Jong PE, Gansevoort RT . Screening for albuminuria identifies individuals at increased renal risk. J Am Soc Nephrol 2009; 20: 852–862.
Kanbay M, Ozkara A, Selcoki Y, Isik B, Turgut F, Bavbek N, Uz E, Akcay A, Yigitoglu R, Covic A . Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int Urol Nephrol 2007; 39: 1227–1233.
Siu YP, Leung KT, Tong MK, Kwan TH . Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006; 47: 51–59.
Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincón A, Arroyo D, Luño J . Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 2010; 5: 1388–1393.
Miao Y, Ottenbros SA, Laverman GD, Brenner BM, Cooper ME, Parving HH, Grobbee DE, Shahinfar S, de Zeeuw D, Lambers Heerspink HJ . Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension 2011; 58: 2–7.
Sánchez-Lozada LG, Soto V, Tapia E, Avila-Casado C, Sautin YY, Nakagawa T, Franco M, Rodríguez-Iturbe B, Johnson RJ . Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol 2008; 295: F1134–F1141.
Kosugi T, Nakayama T, Heinig M, Zhang L, Yuzawa Y, Sanchez-Lozada LG, Roncal C, Johnson RJ, Nakagawa T . Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am J Physiol Renal Physiol 2009; 297: F481–F488.
Price KL, Sautin YY, Long DA, Zhang L, Miyazaki H, Mu W, Endou H, Johnson RJ . Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol 2006; 17: 1791–1795.
Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ . Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001; 38: 1101–1106.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ito, S., Naritomi, H., Ogihara, T. et al. Impact of serum uric acid on renal function and cardiovascular events in hypertensive patients treated with losartan. Hypertens Res 35, 867–873 (2012). https://doi.org/10.1038/hr.2012.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.59
Keywords
This article is cited by
-
The association between serum uric acid and cognitive performance in patients with ischemic stroke is modified by estimated glomerular filtration rate
Scientific Reports (2023)
-
Effects of losartan and enalapril on serum uric acid and GFR in children with proteinuria
Pediatric Nephrology (2021)
-
Impact of long-term potassium supplementation on thiazide diuretic-induced abnormalities of glucose and uric acid metabolisms
Journal of Human Hypertension (2018)
-
Comparison of olmesartan combined with a calcium channel blocker or a diuretic in elderly hypertensive patients (COLM Study): safety and tolerability
Hypertension Research (2015)
-
Chapter 7. Hypertension complicated by other diseases
Hypertension Research (2014)
