
Abstract
Chronic renal ischemia leads to renal fibrosis and atrophy. Activation of the renin-angiotensin–aldosterone system is one of the main mechanisms driving chronic renal ischemic injury. The aim of the present study was to define the effect of aliskiren in chronic ischemia of the kidney. Two-kidney, one-clip mice were used to study chronic renal ischemia. Aliskiren significantly lowered the blood pressure in mice with renal artery constriction (92.1±1.1 vs. 81.0±1.8 mm Hg, P<0.05). Renin expression was significantly increased in ischemic kidneys when treated with aliskiren. In addition, (Pro)renin receptor expression was decreased by aliskiren in ischemic kidneys. Aliskiren treatment significantly increased klotho expression and reduced the expression of fibrogenic cystokines, caspase-3 and Bax in ischemic kidneys. Histological examination revealed that aliskiren significantly reduced the nephrosclerosis score (4.5±1.9 vs. 7.3±0.4, P<0.05). Immunofluorescence staining also showed that aliskiren decreased the deposition of interstitial collagen I in ischemic kidneys. In conclusion, direct renin inhibition significantly reduced renal fibrosis and apoptosis following chronic renal ischemia.
Similar content being viewed by others
Introduction
Preventing the progression of chronic kidney disease (CKD) is vital to ablate the increasing trend of end-stage renal disease.1 To do so, determining the factors mediating the progression of CKD is the key.2, 3 Chronic ischemic nephropathy is thought to be one of the major determining factors in progression of CKD.4 Approximately, 15% of end-stage renal disease is attributed to chronic ischemic nephropathy from renovascular disease.4, 5, 6
Activation of the renin-angiotensin–aldosterone system (RAAS) is one of the main mechanisms of chronic ischemic nephropathy-associated progressive of CKD.7, 8 RAAS activation is considered to have important roles in the renal fibrosis.9 Renal ischemia stimulates the release of renin via afferent arteriole baroreceptors, or by sensing a decreased sodium chloride load to the distal nephron.10 The subsequent production of angiotensin II induces vasoconstriction of both afferent and efferent arterioles, which results in an increase in glomerular pressure and glomerular filtration rate, and in further renal injury.11 These hemodynamic changes occur immediately after ischemia. However, the RAAS alteration in chronic kidney ischemia is less understood.
Increased RAAS activity can act as a multifunctional cytokine such as a growth factor, a profibrogenic cytokine, and can display proinflammatory properties.12, 13 Renin itself has been identified as a mediator of profibrotic effects via binding to specific receptors.14 These non-hemodynamic effects also have a role in renal kidney damage during chronic renal ischemia.
Inhibition of the RAAS is currently one of the most powerful therapies to slow the progression of CKD.14, 15 The angiotensin converting enzyme inhibitor and angiotensin receptor blocker are agents that block the activated RAAS. However, the effectiveness of these treatments is limited due to compensatory increases in plasma renin levels that lead to adjustments in angiotensin production and conversion.16 A direct renin inhibitor, aliskiren, is now available to inhibit angiotensin production directly at its rate-limiting step.17
The goal of the present study was to understand the effect of aliskiren—a direct renin inhibitor—on renal fibrosis and possible mechanisms other than RAAS activation. To address this, we performed an animal study using the two-kidney, one-clip Goldblatt model for chronic ischemic nephropathy.18
methods
Animal model
Four-week-old female B6 mice (National Laboratory Animal Center, Taiwan) were assigned randomly into normal control (n=8) and experimental (n=16) groups. The experimental group received renal artery ligation surgery19 and the control mice received sham surgery. Mice in the experimental group were assigned randomly into two groups: aliskiren treatment group (n=8) and non-treatment group (n=8). Mice of the treatment group received 25 mg kg−1 per day of aliskiren (Novartis, East Hanover, NJ, USA) via osmotic mini-pumps during renal artery ligation surgery, whereas the non-treatment group received no medical treatment for 3 weeks. The dose of aliskiren was determined according to previous studies.20, 21 Renal artery constriction (RAC) was performed by making dorsal, longitudinal incisions to expose the kidneys. The trunk of the left renal artery, which showed visible renal ischemia, was constricted by a vascular clamp (0.4–1.0 mm; S&T Fine Science Tools, North Vancouver, B.C. Canada); the right kidney remained intact. The kidney tissues were harvested 3 weeks after surgery and the kidney weight was measured. The blood pressure of study mice was determined before surgery and 3 weeks post surgery. The mean arterial pressure (2/3 diastolic pressure+1/3 systolic pressure) was determined before RAC and 3 weeks following RAC, with tail-cuff plethysmography.22 All animals were given a normal diet (sodium, 0.25%; potassium, 0.76%) and tap water. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and was approved by the local ethics committee of Chang Gung Memorial Hospital.
Quantitative real-time PCR
Whole kidney extracts (10 mg) were used to isolate total RNA using a commercial kit (RNeasy Kit, Qiagen, Valencia, CA, USA) according to the manufacturer's instructions, including DNase treatment. Total RNA (5 μg) was then reverse transcribed by reverse transcriptase (Bio-Rad, Hercules, CA, USA) with random primers. Real-time PCR was performed in 25 μl SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) containing 0.6 mol l−1 primers and 1 μg cDNA, using an iQ5 real-time PCR detection system (Bio-Rad). The thermal cycling program and PCR primers are listed in Supplementary Table 1. Each PCR reaction was performed in triplicates, and the mean Ct value was used for static analysis. Messenger RNA expression was standardized to β-actin expression levels, followed by normalization to the control group.
Western blotting
Kidney tissues were homogenized and total protein was extracted using a commercial kit as per the manufacturer's instructions (Protein Extraction Kit, Millipore, Billerica, MA, USA). Kidney extracts (30 μg protein per lane) were mixed with a sample loading buffer and separated on 12% SDS-polyacrylamide gel. Proteins were electrotransferred on polyvinylidene fluoride membranes (0.2 μm: Immun-Blot, Bio-Rad). The antibodies used for western blotting are listed in the Supplementary Table 2. The intensity of each band was quantified using the NIH Image software, and the densitometric intensity corresponding to each band was normalized against β-actin expression.
Histopathological examination
Sections of paraffin-embedded specimens were stained with the periodic acid-Schiff reagents and counter-stained with hematoxylin to assess the degree of renal sclerosis. Four random and non-overlapping sections from each kidney tissue were examined under high power field ( × 200) by a pathologist blinded to the study. The nephrosclerosis scores were calculated by adding the scores of glomerulosclerosis, interstitial fibrosis and arterial sclerosis. The severity was graded from 0 to 3+ in each specimen.23, 24
TUNNEL and immunofluorescence staining
Cryostat sections of renal tissue were used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNNEL) and immunofluorescence staining. TUNNEL staining was performed using a MEBSTAIN Apoptosis Kit Direct (MBL International, Woburn, MA, USA) according to the manufacturer's protocol. Sections of renal tissue were incubated with a primary antibody against mouse collagen I at 1:100 dilution, followed by incubation with a fluorescein isothiocyanate-conjugated anti-IgG antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Collagen I expression was determined by assigning an arbitrary grading scale (0 to 5+), based on the kidney interstitial staining intensity. The relative amount of staining was scored semiquantitatively from 0 to 5 as follows: (0) no staining; (1) trace staining; (2) slight staining; (3) moderate staining; (4) intensive staining; and (5) most intense staining. The nucleus was stained with Hoechst stain.
Statistical analysis
All data were expressed as mean±s.e. The results of PCR and western blotting were plotted as the relative fold, as compared with the control group. Data from different study groups were compared using the Wilcoxon–Mann–Whitney test, and P-values of <0.05 were considered statistically significant.
Results
Blood pressure and kidney size
The mean arterial pressure among control and experimental mice was not significantly different at the beginning of the study. Control mice did not exhibit changes in mean arterial pressure during the study period (before: 92.0±1.8 vs. after: 92.2±2.1 mm Hg). The mean arterial pressure increased significantly in mice with RAC (control: 91.3±1.3 vs. RAC+ & aliskiren−: 98.9±1.4 mm Hg, P<0.05). In mice with RAC, the mean arterial pressure decreased significantly after being treated with aliskiren (RAC+ and aliskiren−: 92.1±1.1 vs. RAC+ and aliskiren+: 81.0±1.8 mm Hg, P<0.05; Figure 1a).

Comparisons of blood pressure and kidney weight. Aliskiren significantly decreased mean blood pressure in mice with RAC (92.1±1.1 vs. 81.0±1.8 mm Hg, P<0.05) and reduces the atrophic effect of chronic renal ischemia (aliskiren−: 0.03±0.00; aliskiren+: 0.06±0.01g; P<0.05). (a) The mean arterial pressure (□: before RAC; ▪: 3 weeks after RAC). (b) Weight of kidneys 3 weeks after RAC (*P<0.05).
Kidneys which underwent RAC were significantly smaller than sham-operated mice; aliskiren reduced the atrophic effect of chronic renal ischemia (control: 0.14±0.00; aliskiren−: 0.03±0.00; aliskiren+: 0.06±0.01 gm, P<0.05; Figure 1b).
Effects of aliskiren on kidney RAAS profiles
Chronic renal ischemia decreased renal renin mRNA expression, but did not alter (pro)renin receptor ((P)RR)) and angiotensin II type 1 receptor mRNA expression. AT2 receptor however was significantly decreased in chronic ischemic kidneys. Aliskiren treatment significantly increased renal renin levels and decreased (P)RR mRNA expression in ischemic kidneys. The angiotensin II type 1 and type 2 receptor expression was not significantly altered between kidneys with and without aliskiren treatment (Figure 2).

RAAS profiles of ischemic kidneys analyzed by quantitative PCR. Aliskiren treatment significantly increased renin and decreased the (P)RR expression in ischemic kidneys (*P<0.05).
Aliskiren increased klotho expression
Chronic renal ischemia decreased klotho expression significantly (control vs. kidney with RAC; real-time PCR: 1.00±0.03 vs. 0.05±0.00, P<0.05; western blot: 1.00±0.03 vs. 0.29±0.07, P<0.05; Figure 3). Aliskiren treatment increased klotho mRNA and protein expression in ischemic kidneys significantly (aliskiren+ vs. aliskiren−; real-time PCR: 0.63±0.08 vs. 0.05±0.00, P<0.05; western blot: 0.84±0.04 vs. 0.29±0.07, P<0.05; Figure 3).

Quantitative PCR and western blot results for klotho expression in ischemic kidneys. Aliskiren significantly increased klotho mRNA and protein expression in ischemic kidneys. (a) Quantitative PCR results, (b) western blot results (*P<0.05).
Aliskiren decreased Bax and caspase-3 expression, and reduced cell apoptosis
Kidneys with RAC significantly increased levels of Bax and caspase-3 protein expression compared with normal kidneys (Bax: 2.71±0.07 vs. 1.00±0.01, P<0.05; caspase-3: 5.77±1.01 vs. 1.00±0.13, P<0.05; Figure 4a). Aliskiren treatment significantly decreased Bax and caspase-3 protein expression in ischemic kidneys compared with the non-treatment group (Bax: 1.73±0.09 vs. 2.71±0.07, P<0.05; caspase-3: 2.36±0.10 vs. 5.77±1.01, P<0.05; Figure 4a). TUNNEL staining results showed that the number of TUNNEL-positive cells in renal parenchyma was significantly decreased in RAC mice treated with aliskiren (Figure 4b).

Results of western blotting for Bax and caspase-3, and TUNNEL staining. (a) Aliskiren decreases Bax and caspase-3 protein expression in ischemic kidneys significantly. (b) The number of TUNNEL-positive cells in renal parenchyma was significantly decreased in RAC mice treated with aliskiren (*P<0.05).
Aliskiren decreased transforming growth factor (TGF)-β1 and connective tissue growth factor (CTGF) expression
Chronic renal ischemia increased TGF-β1 and CTGF protein expression when compared with the control mice (TGF-β1: 3.96±0.87 vs. 1.00±0.05, P<0.05; CTGF: 20.22±6.63 vs. 1.00±0.01, P<0.05). Aliskiren treatment decreased TGF-β1 and CTGF protein expression in ischemic kidneys when compared with the non-treated mice (TGF-β1: 1.04±0.28 vs. 3.96±0.87, P<0.05; CTGF: 8.46±2.30 vs. 20.22±6.63, P<0.05; Figure 5).

Western blotting for TGF-β1 and CTGF. Aliskiren significantly reduced intrarenal TGF-β1 and CTGF protein expression in ischemic kidneys (*P<0.05).
Aliskiren decreased collagen I expression, but did not alter collagen III expression
Comparing with the control group, kidneys with RAC showed significantly increased collagen I and III mRNA expression levels (collagen I: 6.03±1.07 vs. 1.00±0.03, P<0.05; collagen III: 3.77±0.53 vs. 1.00±0.11). Aliskiren treatment decreased the collagen I mRNA expression significantly compared with the non-treatment group (aliskiren+ vs. aliskiren−: 2.64±0.28 vs. 6.03±1.07; P<0.05). However, aliskiren treatment did not change the mRNA expression of collagen III and VI in ischemic kidneys (Figure 6).

Quantitative PCR for collagen I, III and IV expression. Aliskiren decreased collagen I mRNA expression in ischemic kidney significantly. Collagen III and IV mRNA expression did not significantly differ between treated and non-treated groups (*P<0.05).
Aliskiren decreased kidney fibrosis in ischemic kidney
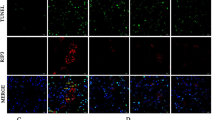
Histochemical staining revealed that chronic renal ischemia caused by RAC led to severe nephrosclerosis and inflammation. Furthermore, even with aliskiren treatment, kidneys with RAC showed prominent kidney fibrosis when compared with sham-operated mice (Figure 7a). Immunostaining revealed that interstitial collagen I deposition was minimal in normal control kidneys (score: 0). In contrast, the intensity of interstitial collagen I deposition in ischemic kidney without aliskiren treatment was stronger (score: 5+). However, treatment with aliskiren reduced the intensity of interstitial collagen I deposition in ischemic kidneys (score: 2+; Figure 7a). The nephrosclerosis scores of study mice were plotted in Figure 7b. The severity of nephrosclerosis was significantly decreased in kidneys with aliskiren treatment compared with those without treatment.

Representative results of pathological staining, and plots of nephrosclerosis scores. (a) Results of periodic acid-Schiff and immunofluorescence staining. Aliskiren significantly reduced renal fibrosis in chronic ischemic kidneys. In the normal control mice, only minimal kidney fibrosis and collagen I staining (score: 0) are noted. Experimental mice without aliskiren treatment are associated with marked collagen I staining (score: 5+). Experimental mice with aliskiren treatment are associated with a reduction in the collagen I staining (score: 2+). (b) Plots of nephrosclerosis scores. The nephrosclerosis scores of normal control and ischemic kidneys with and without aliskiren treatment were 0.5±0.2, 4.5±0.2 and 7.3±0.4, respectively (*P<0.05).
Discussion
Chronic renal ischemia is different from acute ischemia injury.25, 26 Although chronic renal ischemia is more relevant to clinical CKD, it is not as widely studied as acute ischemia injury. Our study revealed that aliskiren treatment attenuates the effect of chronic ischemia on klotho expression, apoptosis and renal fibrosis. Aliskiren treatment reduced blood pressure and attenuated renal renin decrement. Our study also revealed that intrarenal renin expression was decreased in chronic ischemic kidneys, which is in agreement to a previous study in rats.27 The decreased renin expression in chronic ischemic kidneys may come from local chronic ischemia that may destroy the renin-producing cells.28
In vivo, (P)RR transgenic rats develop glomerulosclerosis and hypertension in the absence of changes to renin or angiotensin levels.29 Various animal models demonstrated that inhibition of pro-renin binding to the (P)RR can prevent or even abolish the development of cardiac fibrosis and diabetic nephropathy.30 In our study, aliskiren significantly decreased renal (P)RR mRNA expression in chronic ischemia. The observed effect may suggest that decreasing renal (P)RR expression has an important role in the protective renal effect of aliskiren.
The klotho is an anti-aging gene that is predominantly expressed in the kidney.31 Renal klotho expression was found to decrease in spontaneously hypertensive rats, 5/6 nephrectomized rats, non-insulin-dependent diabetes mellitus rats and in ischemia–reperfusion injury models.32 In humans, klotho expression also decreases in CKD patients,33 and also has protective renal effects by protecting cells/tissues from oxidative stress and apoptosis.29 In our study, we demonstrated that direct renin inhibition significantly increased klotho expression in vitro and in vivo. The increased expression of klotho gene might further protect chronic ischemic renal injury in animals undergoing aliskiren treatment.
Our results revealed that direct renin inhibition could effectively alleviate the kidney fibrosis caused by chronic kidney ischemia. Aliskiren is designed to block the enzymatic site of human renin. The difference in the amino acid sequence and stereo structure of human and mouse renin could alter the pharmacodynamics of aliskiren. Animals expressing human renin gene have often been used as animal models in studies for aliskiren.34, 35 However, recent reports showed that aliskiren still had renal protective effects in the animal models without expressing human renin.36, 37 Our study also showed the similar results. In addition, our data demonstrated that the intrarenal renin expression decreased when chronic renal ischemic injury advanced. Whether mechanisms other than direct renin inhibition had roles in the renal protective effects of aliskiren needs further studies to define.
There are several limitations in our study. The number of study animals is small, which could limit the reliability of the statistical analysis in this study.
In conclusion, direct renin inhibition with aliskiren could increase klotho expression, reduce fibrogenic cytokine production and cell apoptosis, and attenuate the kidney fibrosis in an animal model of chronic ischemic renal injury.
References
Foley RN, Collins AJ . End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol 2007; 18: 2644–2648.
Yang WC, Hwang SJ . Incidence, prevalence and mortality trends of dialysis end-stage renal disease in Taiwan from 1990 to 2001: the impact of national health insurance. Nephrol Dial Transplant 2008; 23: 3977–3982.
Levin A, Djurdjev O, Beaulieu M, Er L . Variability and risk factors for kidney disease progression and death following attainment of stage 4 CKD in a referred cohort. Am J Kidney Dis 2008; 52: 661–671.
Masuo K, Lambert GW, Esler MD, Rakugi H, Ogihara T, Schlaich MP . The role of sympathetic nervous activity in renal injury and end-stage renal disease. Hypertens Res 2010; 33: 521–528.
Preston RA, Epstein M . Ischemic renal disease: an emerging cause of chronic renal failure and end-stage renal disease. J Hypertens 1997; 15: 1365–1377.
Lee CC, Sun CY, Wu MS . Long-term modality-related mortality analysis in incident dialysis patients. Perit Dial Int 2009; 29: 182–190.
Siddiqi L, Joles JA, Grassi G, Blankestijn PJ . Is kidney ischemia the central mechanism in parallel activation of the renin and sympathetic system? J Hypertens 2009; 27: 1341–1349.
Rea ME, Dunlap ME . Renal hemodynamics in heart failure: implications for treatment. Curr Opin Nephrol Hypertens 2008; 17: 87–92.
Zeisberg M, Neilson EG . Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 2010; 21: 1819–1834.
Wang X, Breaks J, Loutzenhiser K, Loutzenhiser R . Effects of inhibition of the Na+/K+/2Cl− cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am J Physiol Renal Physiol 2007; 292: F999–F1006.
Bader M, Ganten D . Regulation of renin: new evidence from cultured cells and genetically modified mice. J Mol Med 2000; 78: 130–139.
Berl T . Review: renal protection by inhibition of the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst 2009; 10: 1–8.
Lijnen P, Petrov V . Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol 2000; 32: 865–879.
Ruster C, Wolf G . Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 2006; 17: 2985–2991.
Campbell RC, Ruggenenti P, Remuzzi G . Halting the progression of chronic nephropathy. J Am Soc Nephrol 2002; 13 (Suppl 3): S190–S195.
Hollenberg NK . Direct renin inhibition and the kidney. Nat Rev Nephrol 2010; 6: 49–55.
Schmieder RE, Philipp T, Guerediaga J, Gorostidi M, Smith B, Weissbach N, Maboudian M, Botha J, van Ingen H . Long-term antihypertensive efficacy and safety of the oral direct renin inhibitor aliskiren: a 12-month randomized, double-blind comparator trial with hydrochlorothiazide. Circulation 2009; 119: 417–425.
Muller DN, Klanke B, Feldt S, Cordasic N, Hartner A, Schmieder RE, Luft FC, Hilgers KF . (Pro)renin receptor peptide inhibitor ‘handle-region’ peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension 2008; 51: 676–681.
Armenia A, Sattar MA, Abdullah NA, Khan MA, Johns EJ . Functional subtypes of renal alpha1-adrenoceptor in diabetic and non-diabetic 2K1C Goldblatt renovascular hypertension. Acta Pharmacol Sin 2008; 29: 564–572.
Dong YF, Liu L, Lai ZF, Yamamoto E, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, Nako H, Ogawa H, Kim-Mitsuyama S . Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia 2010; 53: 180–191.
Ino J, Kojima C, Osaka M, Nitta K, Yoshida M . Dynamic observation of mechanically-injured mouse femoral artery reveals an antiinflammatory effect of renin inhibitor. Arterioscler Thromb Vasc Biol 2009; 29: 1858–1863.
Buñag RD . Validation in awake rats of a tail-cuff method for measuring systolic pressure. J Appl Physiol 1973; 34: 279–282.
Ono H, Ono Y, Frohlich ED . ACE inhibition prevents and reverses L-NAME exacerbated nephrosclerosis in spontaneously hypertensive rats. Hypertension 1996; 27: 176–183.
Vikse BE, Bostad L, Aasarød K, Lysebo DE, Iversen BM . Prognostic factors in mesangioproliferative glomerulonephritis. Nephrol Dial Transplant 2002; 17: 1603–1613.
Shanley PF . The pathology of chronic renal ischemia. Semin Nephrol 1996; 16: 21–32.
Kellerman PS . Cellular and metabolic consequences of chronic ischemia on kidney function. Semin Nephrol 1996; 16: 33–42.
el-Dahr SS, Dipp S, Guan S, Navar LG . Renin, angiotensinogen, and kallikrein gene expression in two-kidney Goldblatt hypertensive rats. Am J Hypertens 1993; 6: 914–919.
Krebs C, Hamming I, Sadaghiani S, Steinmetz OM, Meyer-Schwesinger C, Fehr S, Stahl RA, Garrelds IM, Danser AH, van Goor H, Contrepas A, Nguyen G, Wenzel U . Antihypertensive therapy upregulates renin and (pro)renin receptor in the clipped kidney of Goldblatt hypertensive rats. Kidney Int 2007; 72: 725–730.
Danser AH . (Pro)renin receptors: are they biologically relevant? Curr Opin Nephrol Hypertens 2009; 18: 74–78.
Feldman DL, Jin L, Xuan H, Contrepas A, Zhou Y, Webb RL, Mueller DN, Feldt S, Cumin F, Maniara W, Persohn E, Schuetz H, Jan Danser AH, Nguyen G . Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension 2008; 52: 130–136.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI . Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390: 45–51.
Wang Y, Sun Z . Current understanding of klotho. Ageing Res Rev 2009; 8: 43–51.
Koh N, Fujimori T, Nishiguchi S, Tamori A, Shiomi S, Nakatani T, Sugimura K, Kishimoto T, Kinoshita S, Kuroki T, Nabeshima Y . Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 2001; 280: 1015–1020.
Pilz B, Shagdarsuren E, Wellner M, Fiebeler A, Dechend R, Gratze P, Meiners S, Feldman DL, Webb RL, Garrelds IM, Jan Danser AH, Luft FC, Müller DN . Aliskiren, a human renin inhibitor, ameliorates cardiac and renal damage in double-transgenic rats. Hypertension 2005; 46: 569–576.
Schmerbach K, Pfab T, Zhao Y, Culman J, Mueller S, Villringer A, Muller DN, Hocher B, Unger T, Thoene-Reineke C . Effects of aliskiren on stroke in rats expressing human renin and angiotensinogen genes. PLoS One 2010; 5: e15052.
Dong YF, Liu L, Lai ZF, Yamamoto E, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, Nako H, Ogawa H, Kim-Mitsuyama S . Aliskiren enhances protective effects of valsartan against type 2 diabetic nephropathy in mice. J Hypertens 2010; 28: 1554–1565.
Gross O, Girgert R, Rubel D, Temme J, Theissen S, Müller GA . Renal protective effects of aliskiren beyond its antihypertensive property in a mouse model of progressive fibrosis. Am J Hypertens 2011; 24: 355–361.
Acknowledgements
The aliskiren powder was kindly provided by Novartis. We thank the staff of Keelung Chang Gung Memorial Hospital Research Center for their assistance with this investigation. This work was funded by a grant from the National Science Council of Taiwan (NMRPG266023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Hypertension Research website
Supplementary information
Rights and permissions
About this article
Cite this article
Sun, CY., Cherng, WJ., Jian, HZ. et al. Aliskiren reduced renal fibrosis in mice with chronic ischemic kidney injury—beyond the direct renin inhibition. Hypertens Res 35, 304–311 (2012). https://doi.org/10.1038/hr.2011.181
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2011.181
Keywords
This article is cited by
-
Soluble Klotho is not independently associated with cardiovascular disease in a population of dialysis patients
BMC Nephrology (2014)
-
Direct renin inhibitor prevents and ameliorates insulin resistance, aortic endothelial dysfunction and vascular remodeling in fructose-fed hypertensive rats
Hypertension Research (2013)
-
Mechanistic insights into the antifibrotic activity of aliskiren in the kidney
Hypertension Research (2012)
