
Abstract
Cardiovascular functions, including blood pressure and vascular functions, show diurnal oscillation. Circadian variations have been clearly shown in the occurrence of cardiovascular events such as acute myocardial infarction. Circadian rhythm strongly influences human biology and pathology. The identification and characterization of mammalian clock genes revealed that they are expressed almost everywhere throughout the body in a circadian manner. In contrast to the central clock in the suprachiasmatic nucleus (SCN), the clock in each tissue or cell is designated as a peripheral clock. It is now accepted that peripheral clocks have their own roles specific to each peripheral organ by regulating the expression of clock-controlled genes (CCGs), although the oscillation mechanisms of the peripheral clock are similar to that of the SCN. However, little was known about how the peripheral clock in the vasculature contributes to the process of cardiovascular disorders. The biological clock allows each organ or cell to anticipate and prepare for changes in external stimuli. Recent evidence obtained using genetically engineered mice with disrupted circadian rhythm showed a novel function of the internal clock in the pathogenesis of endothelial dysfunction, hypertension and hemostasis. Loss of synchronization between the central and peripheral clock also contributes to the pathogenesis of cardiovascular diseases, as restoration of clock homeostasis could prevent disease progression. Identification of CCGs in each organ, as well as discovery of tools to manipulate the phase of each biological clock, will be of great help in establishing a novel chronotherapeutic approach to the prevention and treatment of cardiovascular disorders.
Similar content being viewed by others

Introduction
It is well known that some cardiovascular physiological functions, such as heart rate (HR) and blood pressure (BP), show apparent circadian variation. In addition, many cardiovascular disorders occur in a circadian manner. For example, acute myocardial infarction (AMI) and cerebral infarction most often occur in the early morning whereas subarachnoid hemorrhage and a subtype of atrial fibrillation are usually observed in the afternoon. The diurnal variation in cardiovascular events is believed to be the consequence of both external and internal biological clock rhythms. Most of these disorders, once they happen, can be fatal or induce severe damage; therefore, it is important to elucidate the precise mechanism of the onset of such diseases to establish a preventive strategy. In this article, we reviewed the role of the molecular clock in the pathogenesis of vascular diseases.
Molecular clock in mammalian cells
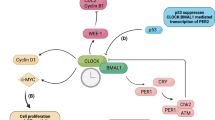
Accumulating evidence has elucidated the molecular mechanisms of the circadian clock.1, 2, 3, 4 Several positive and negative feedback loops exist in the biological clock at transcriptional and post-translational levels. Among them, the core negative feedback loop comprises positive limb (CLOCK, NPAS2, BMAL1 and CLIF/BMAL2) proteins and negative limb (three period (PER1, PER2 and PER3) and two cryptochrome (CRY1 and CRY2)) proteins. Most are basic helix-loop-helix/per-arnt-sim domains containing transcription factors. CLOCK or NPAS2 forms a heterodimer with BMAL1 or CLIF/BMAL2 and binds to the E-box element with CACGTG sequences upstream of the per or cry gene.2 They enhance the transcription of per and cry, and the PER protein forms a complex with the CRY protein and inhibits CLOCK/BMAL-mediated transcription of the per or cry gene itself, and therefore resulting in a negative feedback loop (Figure 1). PER proteins are also phosphorylated with serine-threonine kinase casein kinase 1-ɛ and degraded by the proteasomal pathway.5 Thus, post-translational mechanisms, including phosphorylation and ubiquitination, also control the timing of the circadian clock.6 In contrast to the ubiquitous expression of BMAL1, CLIF/BMAL2 is mainly expressed in vascular endothelial cells.7 However, little is known about the redundancy or the dynamic function of BMALs in the vasculature, although CLIF/BMAL2 was shown to have a higher affinity to PER2 than BMAL1.8

The heterodimer of CLOCK and BMAL1 binds to the E-box elements upstream of period (per), cryptochrome (cry) and nuclear receptor Rev-erbα promoters. PER protein accumulates in the cytoplasm and translocates into the nucleus, forming a complex with CRY proteins, and then inhibits CLOCK-BMAL1-dependent transcription. Rev-erbα protein accumulates quickly and inhibits BMAL1 transcription, resulting in the oscillation of bmal1 gene expression. CLOCK/BMAL1 heterodimer also binds to the E-box of target genes, designated as clock-controlled genes (CCGs). The heterodimer also transactivates proline- and acid-rich basic leucine zipper transcription factors, dbp, hlf and tef. These transcription factors in turn induce the circadian expression of CCGs.
In addition to this core feedback loop, the nuclear receptor REV-ERBα is also transactivated by the CLOCK/BMAL1 heterodimer. The REV-ERBα protein represses bmal1 transcription, which is essential for circadian bmal1 expression. Another feedback loop includes the basic helix-loop-helix domain containing transcription factors, deleted in esophageal cancer (dec1 and dec2). The heterodimer of CLOCK and BMAL1 binds to the E-box upstream of dec1 and dec2, and activates their transcription. DEC proteins in turn repress the transcriptional activity of CLOCK/BMAL1, thus forming another negative feedback loop. The CLOCK/BMAL1 heterodimer binds to the E-box upstream of not only the per or cry gene, but also to other target genes designated as clock-controlled genes (CCGs). The CCGs include arginine vasopressin, wee1 or other target genes, and mediate the rhythmicity of the biological clock and account for the circadian variation in humoral or metabolic functions. The CCGs also comprise three proline- and acid-rich (PAR) basic leucine zipper transcription factors: D-element binding proteins (dbp), hepatic leukemia factor (hlf) and thyrotrophic embryonic factor (tef).4 In addition to CLOCK/BMAL1, PAR transcription factors also induce the circadian expression of CCGs and therefore act as mediators or amplifiers of CLOCK/BMAL-induced CCG expression. The induction of CCG expression is antagonized by another basic helix-loop-helix transcription factor, E4BP4,9 which is induced by REV-ERBα. The phase of the three PAR transcription factors are antiphase to that of E4BP4, resulting in the circadian expression of CCGs.
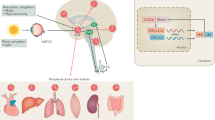
The center of the biological clock, that is, the central clock, exists in the suprachiasmatic nucleus (SCN) in the hypothalamus.3 The central clock regulates physiological functions through the autonomic nervous system, humoral mediators or unknown factors. The phases of the internal clock can be entrained by external stimuli. Zeitgebers (timekeepers) are factors that could reset the rhythm. Clock genes express in a circadian manner in SCN, and light is the main zeitgeber for the central clock and can reset the phase of its rhythm. In addition to the central clock, circadian expression of clock genes can be detected in each peripheral organ or cell, suggesting that each organ has its own internal clock. This clock system is called the peripheral clock in contrast to the central clock in the SCN (Figure 2). The molecular mechanism of the peripheral clock is considered to be similar to that of the central clock.10, 11 The central clock synchronizes each of the peripheral clocks within the body;1 however, little is known about how peripheral clocks are regulated by the central clock. In contrast to the central clock, the phase of the peripheral clock cannot be entrained with light; thus, the phases of each peripheral clock seem to be synchronized by neuronal or other unknown humoral factors derived from the SCN. Finding the appropriate zeitgeber for each organ will help in not only understanding the clock system, but also in establishing a novel type of therapeutic approach, named chronotherapy.

The center of the biological clock (central clock) is located in suprachiasmatic nucleus (SCN) in the hypothalamus. Each organ or cell, including the aorta, vascular endothelial cells and vascular smooth muscle cells (VSMC), also has circadian expression of clock genes and is designated as the peripheral clock. Circadian expression of clock-controlled genes (CCGs) is in part regulated directly by the central clock (direct pathway). In addition, peripheral clocks in cardiovascular tissues or cells are also stimulated and synchronized by the central clock and regulate diurnal expression of CCGs.
Molecular/peripheral clock in vasculature
The existence of a peripheral clock system in each organ or cell was shown using in vitro cultured fibroblasts.10 Balsalobre et al.10 stimulated fibroblasts with 50% serum for a short time and observed the circadian oscillation of clock gene expression. A single cell in culture has its own oscillation rhythm, whereas cell populations in in vitro culture are usually arrhythmic because of the asynchronous circadian rhythm among cells.12 However, once a phase-aligning stimulus such as 50% serum is applied, they start to show uniform circadian rhythm.13
Diurnal variation in clock genes was also reported in cardiac organs including the heart, aorta and kidney.14, 15 A study based on microarray analysis revealed that approximately 8–10% of genes show circadian expression in the heart and liver; however, most of these genes are organ specific.16 Therefore, in addition to the central clock in the SCN, a peripheral clock in each organ seems to regulate tissue-specific physiological functions, and identification of peripheral CCG will greatly help in understanding the role of the biological clock in cardiovascular organs.17
To prove the existence of an intrinsic clock system in cardiovascular tissues, we studied the clock gene expression of in vitro cultured vascular endothelial cells and confirmed the circadian clock gene expression.18 We also searched for CCGs in vascular endothelial cells and identified circadian expression of thrombomodulin, a membrane protein with anticoagulant activity. Vascular smooth muscle cells also possess an intrinsic biological clock. In addition to serum shock, angiotensin II or retinoic acid also induced circadian clock gene expression, suggesting that they can function as a zeitgeber.15, 19 Chalmers et al.20 identified that the tissue inhibitor of metalloproteinase 1 and 3, collagen 3a1, transgelin1 (sm22α) and calponin1 show circadian expression in smooth muscle cell line (Movas-1). Several vascular functions have been shown to show circadian rhythm, including endothelium-dependent vasodilatory function.21, 22 In human subjects, endothelial function measured by flow-mediated dilation was shown to have circadian oscillation with lower function in the morning.23
Recent evidence has illuminated the roles of the molecular clock in endothelial functions. Mice with the Per2 mutation produced lesser amounts of nitric oxide and vasodilatory prostaglandins and more cyclooxygenase-1-derived vasoconstrictors than the wild type, resulting in impaired endothelium-dependent relaxation in response to acetylcholine.24 Endothelial dysfunction was also observed in mice with Bmal1 knockout and CLOCKmut.25 Akt signaling and nitric oxide production were reduced in Bmal1 knockout arteries, and these arteries became more susceptible to thrombosis formation. The release of hematopoietic stem cells or endothelial progenitor cells (EPCs) from bone marrow is regulated by circadian rhythm.26 In a diabetic state, a disrupted peripheral clock caused by bone marrow neuropathy impaired circadian release of EPCs from bone marrow and exacerbated diabetic retinopathy.27 Per2 mutant mice also had impaired EPC mobilization function.28 EPC mobilization in response to ischemia or vascular endothelial growth factor stimulation was reduced in Per2 mutant mice compared with wild-type mice. EPCs from Per2 mutant mice showed greater senescence together with Akt activation and impaired angiogenesis in a hind-limb ischemia model. Transplantation of wild-type bone marrow into Per2 mutant mice prevented autoamputation in Per2 mutant mice. Both Bmal1 knockout and Per2 mutant mice had endothelial dysfunction; however, the opposite effect was observed with respect to Akt activation. This may be related to the different roles of these clock genes in the core loop formation; that is, Bmal-1 is a positive limb protein whereas Per2 works as a negative limb protein. Senescence also affects the biological clock function. Kunieda et al.29 revealed that circadian expression of clock genes are attenuated in senescent human smooth muscle cells. Telomere shortening and impaired cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) activation accounted for the loss of circadian rhythmicity in senescent cells, as the introduction of telomerase or restoration of CREB affected a complete recovery of the circadian rhythm.
BP/hypertension and the molecular clock
In the normal subjects, BP declines during night time, begins to rise in early hours of the morning and reaches near peak or peak values at mid-morning.30, 31 A number of factors can influence the diurnal variation in BP, including the autonomic nervous system,32 vasoactive intestinal peptide,33 plasma renin activity,34 aldosterone35 and plasma atrial natriuretic peptide.36 It is well known that sympathetic activity as well as renin–angiotensin–aldosterone activity peaks in the morning.32, 37 BP is also affected by external factors such as physical activity, emotional state, eating and the sleep/wake cycle. Results of a study in humans indicated that disharmony in the circadian rhythm can cause hypertension.38 Human subjects kept under a protocol of circadian misalignment with behavioral cycle of 28 h instead of 24 h showed mild but significant hypertension.
A decade ago, Janssen et al.39 studied the role of the internal clock in the rhythm of BP. Lesioning of the rat SCN abolished the circadian rhythm of BP and HR without affecting the 24-h cycle of locomoter activities. Recent evidence has provided much deeper insights into the role of the molecular clock in BP regulation. Global deletion of Bmal1 completely abolished the diurnal variation in BP.40 Bmal1 mutant mice also show hypotension together with reduced production of catecholamines. Global Per2 mutant mice also show lower BP.24 In contrast, the endothelial-specific deletion of Bmal1 did not affect the variation in BP, suggesting that the peripheral clock in endothelial cells does not solely induce diurnal BP rhythm.41 A genetic association study showed that a single-nucleotide polymorphism within the bmal1 promoter is associated with hypertension and type II diabetes,42 providing support that the molecular clock is involved in the pathogenesis of metabolic disorders. Recent evidence supported the contribution of peroxisome proliferator-activated receptor-γ (PPARγ) in the clock system. PPARγ binds to the promoter upstream of bmal1 and induces its transcription.43 The expression of PPARγ also showed circadian oscillation in the aorta, and an endothelial- or vascular smooth muscle cell-specific deletion of PPARγ attenuated the BP variation together with reduced catecholamine production.
Plasma aldosterone concentration has a diurnal variation with the peak during night hours.35 One of the adrenal enzymes involved in aldosterone production, type VI 3β-hydroxyl-steroid dehydrogenase (Hsd3b6), shows circadian expression in normal subjects. However, Cry1/2-null mice had a constitutive high expression of Hsd3b6 together with overproduction of aldosterone from adrenal glands, which resulted in salt-sensitive hypertension in Cry1/2-null mice.44 Aldosterone regulates the expression of the alpha-subunit of the epithelial sodium channel (αENaC) mRNA through the Per1-mediated pathway.45 αENaC is known to affect systemic BP; therefore, these findings suggest a novel function of the molecular clock during the pathogenesis of hypertension.
AMI and the circadian clock
Beginning a few decades ago, it became well known that AMI or thrombotic events such as pulmonary embolism frequently occur in the early morning.46, 47 As these disorders can be fatal, elucidating the mechanisms of circadian onset of cardiovascular disorders will help not only for a better understanding of their pathogenesis but also for establishing preventive strategies.48 In this section, we discuss how the biological clock contributes to the onset of thromboembolic events.
Diurnal activation of the autonomic nervous system seems to contribute to the circadian onset of cardiovascular events. A morning increase in ischemic events was not observed in patients with autonomic nervous dysfunction induced by diabetes.49 In addition, patients receiving β-blockers did not show morning increase of ischemic heart attacks.50 Several cardiovascular and hematologic functions are related to the circadian onset of cardiovascular events, including BP, HR, coronary blood flow, platelet function, blood coagulability and fibrinolytic activity.48 In the early morning, BP and HR increase and enhance the demand for oxygen by the heart.51 In contrast, the vascular tone of coronary arteries increases and, therefore, coronary blood flow decreases in the morning,52 resulting in a mismatch of oxygen demand and supply during this period. Coronary segments with endothelial dysfunction show circadian vasomoter activity, whereas segments with normal endothelial function did not show circadian variations, suggesting a potential protective role of endothelial function in coronary events.53 Moreover, both platelet aggregation and blood coagulability increase,54 whereas fibrinolytic activity decreases in the morning. These hypercoagulability and hypofibrinolytic activities also elicit the morning onset of thromboembolic events.
Not only platelet aggregation activity, but also the number of circulating platelets have circadian oscillation.55, 56 Platelets are activated by catecholamines, which are secreted from the autonomic nervous system. However, it is not clear whether the peripheral clock directly affects platelet function, as no surface markers characteristic of platelet activation have been shown thus far to show circadian expression.55
High concentration of coagulation factor VII is considered to be a risk factor for coronary artery diseases.57 Circadian oscillation has been shown not only in the factor VII level in blood, but also in the levels of fibrinogen, prothrombin, factor VIII and tissue factor pathway inhibitor, a direct inhibitor of the FXa/TF/FVIIa complex.58, 59 Microparticles from endothelium induce coagulation through the tissue factor-mediated pathway.60 A recent report by Madden et al.61 showed that the number of vascular cell adhesion molecule-1-positive microparticles in human plasma had a significant diurnal variation with a peak at 9 in the morning. These findings support the presence of hypercoagulability in the morning hours.
Fibrinolytic activity was also shown to have circadian variation with a peak in the afternoon and trough in the early morning, which is an antiphase to that of coagulation activity.62, 63, 64 The level of plasmin-plasmin inhibitor complex, a marker of intravascular plasmin generation, decreases in the morning. Because of the morning decrease in fibrinolytic activity, recovery of patency of occluded coronary vessels by tissue plasminogen activator therapy for AMI treatment is more difficult in the morning hours.65 The level of tissue plasminogen activator inhibitor-1 (PAI-1), which regulates the activity of tissue plasminogen activator, mainly determines fibrinolytic activity. High concentration of PAI-1 or tissue plasminogen activator can become a risk factor for the occurrence of a first AMI.66 There is circadian oscillation in the concentration and activity of PAI-1 with a morning peak, resulting in reduced tissue plasminogen activator activity during that period.67, 68 All these data support the notion that circadian oscillation of PAI-1 activity significantly contributes to the formation of a diurnal variation in fibrinolytic function. The homeostasis of the coagulation cascade is achieved by the balance between coagulation activity and fibrinolytic activity. Activation of coagulation is normally accompanied by an increase in fibrinolytic activity. Therefore, the mismatch of these two cascades also elicits the morning onset of cardiovascular events.
The mechanisms of the diurnal variation in PAI-1 activity have been well studied. We and other groups analyzed the roles of the molecular clock in circadian PAI-1 activation.7, 69 PAI-1 mRNA and protein levels clearly reflect a circadian rhythm in the heart and aorta with a peak expression in the evening. The phase of circadian PAI-1 expression in mice is antiphase to that of the humans, as humans are diurnal whereas rodents are nocturnal. Therefore, PAI-1 expression in rodents also accounts for the human circadian oscillation. We have shown that CLIF/BMAL2 forms a heterodimer with CLOCK and binds to the E-boxes upstream of the pai-1 gene and transactivates its expression.7 The heterodimer of CLOCK/BMAL1 also activates the PAI-1 promoter.70 Oishi et al.71 showed that a ketogenic diet induces the phase shift of peripheral clock gene expression including PAI-1, suggesting that PAI-1 expression is regulated by the peripheral clock. Westgate et al.41 studied the susceptibility to thrombotic events using a mouse photochemical injury model and observed a diurnal variation in thrombogenicity in this in vivo model. CLOCKmut mice have lost this dynamic variation. Surprisingly, the endothelial-specific deletion of the Bmal1 gene (Bmal1fx/fxCreTek) also abolished the circadian oscillation of thrombogenic events; however, diurnal variation in systemic PAI-1 activity was sustained in this mouse model. This finding suggests that the peripheral clock within endothelial cells contributes to prevention of thrombosis through mechanisms other than those affecting systemic PAI-1 activity.
Thrombomodulin has an opposite effect to that of PAI-1 in terms of the coagulation cascade; that is, thrombomodulin inhibits thrombin activation and also activates protein C.72, 73, 74 We revealed that thrombomodulin is expressed with a circadian oscillation in vascular endothelial cells.18 The phase of circadian thrombomodulin expression is similar to that of PAI-1 with a peak in the morning. On the basis of these findings, we can raise the hypothesis that circadian expression of thrombomodulin may be beneficial in protecting endothelium from diurnal thrombogenic activation induced by PAI-1 expression. Further studies are required to fully elucidate the role of circadian thrombomodulin expression in cardiovascular events.
Roles of the peripheral clock in cardiovascular diseases
The central questions related to the molecular clock and cardiovascular diseases are whether the biological clock is affected in cardiovascular disorders, and, in turn, whether impairment of the molecular clock induces the progression of these diseases. The impairment of the peripheral clock in pathology has already been shown in several disease models. Young et al.75 showed that the phase of circadian rhythm of core clock genes, such as bmal1, per2 and hlf, was advanced 3 h in diabetic rats. In addition, in rat heart with pressure-overload hypertrophy, the rhythmic expression of PAR transcription factors (dbp and hlf) and anp was markedly reduced.76 Myocardial ischemia/reperfusion was also shown to affect the circadian clock system. Clock gene oscillations were rapidly diminished in the ischemia/reperfusion area of the heart whereas they were not affected in nonischemic regions. E4BP4 antagonizes the transcriptional activity of PAR family members, such as DBP, HLF and TEF. At the ischemia/reperfusion site of the heart, E4BP4 expression was strongly induced, resulting in the suppression of circadian pdk4 and ucp3 expression.77 Moreover, aging and hypertension were also known to affect the internal circadian rhythm.78, 79
Several studies have addressed the second question, which is whether an impaired circadian clock affects disease progression. Penev et al.80, 81 repeated phase shifts of the light/dark cycle in cardiomyopathic hamsters and found that disruption of rhythmicity strikingly enhanced disease progression and resulted in shortened longevity. Martino et al.82 also analyzed the effect of impaired rhythm in cardiac hypertrophy. They performed transverse aortic constriction surgery in a murine model of pressure overload cardiac hypertrophy, and kept the mice in a rhythm-disruptive 20-h (light/dark 10:10) or normal 24-h (light/dark 12:12) environment after transverse aortic constriction surgery. Rhythm-disturbed transverse aortic constriction animals showed decreased left ventricular systolic function together with increased perivascular and interstitial fibrosis. Decreased left ventricular function was recovered when the mice were kept under conditions of a normal 24-h rhythm.
Martino et al.82 also performed an elegant study using hamsters with a point mutation in the circadian regulatory gene casein kinase-1ɛ as a heterozygote, termed the +/tau mutation. The +/tau heterozygous animals had a reduced circadian period of 22 h with disrupted behavior rhythmicity, and they developed cardiomyopathy and extensive cardiac fibrosis, resulting in death at a young age. However, when these mutant animals were maintained under conditions of their own rhythm period (22 h), the progression of the cardiac disorders was reversed. Ablation of the SCN at a young age also rescued the cardiac phenotype. There exist two clock systems with different periods in +/tau heterozygotes, as their peripheral clock is controlled by the intrinsic 22-h clock as well as the 24-h cycle from the SCN. Under a 22-h light/dark cycle or an SCN-lesioned condition, the discrepancy between the central and peripheral clock disappeared together with rescue of the cardiac pathology. These results raise the hypothesis that it is not disruption of the peripheral clock but disharmony between the external and internal clock or between the central and peripheral clock that elicited cardiovascular disorders. Therefore, loss of synchronization between the central and peripheral clock could elicit progression of the disease.
In healthy subjects, the peripheral clock seems to be beneficial for anticipation and preparation for external stimuli such as BP rise in the morning. It may help the organ to respond rapidly and easily to the environmental change at each time of the day. Mice had a diurnal variation in BP with a peak in the evening. In mouse cardiomyocytes, the expression of cardioprotective gene anp also results in a diurnal variation with a peak in dark phase, which is consistent with high BP periods.76
Discrepancy between the two clock systems could occur among peripheral tissues as well. In the aorta, the phase of circadian Per2 expression is distinct from the phase in SCN, suggesting that the timing of clock rhythm is determined by each peripheral organ.83 Davidson et al.84 reported that the time phases of circadian rhythm in arteries and veins vary significantly according to the anatomical location. The role of the peripheral clock in arteries may be different from that in veins.
In the acute phase of myocardial infarction, the phase of the circadian clock in the ischemic heart differs from that in the nonischemic area.77 This discrepancy could elicit the incidence of myocardial arrhythmia. These losses of synchronization of circadian rhythms between each organ or tissue may occur more frequently than we have expected.
Resynchronization of the peripheral circadian clock with the environment or within each peripheral organ can become a potential target for establishing a novel preventive strategy or treatment for cardiovascular diseases. Although angiotensin II, endothelin or prostaglandin E2 is known to modify the circadian rhythm,15, 85, 86 we must identify the appropriate zeitgebers (timekeepers) to reset or resynchronize the phase of each clock system without directly affecting tissue homeostasis. No direct evidence has been reported that catecholamine or nutrients (glucose or fatty acids) could affect the phase of circadian clock.87 A recent report revealed that PPARγ induces Bmal1 expression in cardiovascular organs,43 suggesting that thiazolidinediones, an agonist of PPARγ, may become a potential tool for manipulating the clock system.
Conclusion
Each cardiovascular organ or cell has its own peripheral clock together with input from the SCN central clock. These peripheral clocks seem to have an important role in the prevention of cardiovascular disorders. Identification of CCGs in each organ will provide significant insights for an understanding of the precise roles of the peripheral clock. Synchronization of clock cycles between the central and peripheral clock, or among peripheral clocks in different organs, is also critical for normal health and homeostasis. Failure to harmonize the central and peripheral clock or internal and external rhythm could result in progression of cardiovascular disorders. Discovery of an appropriate zeitgeber or a small compound that could manipulate the phase of each peripheral clock is required to establish chronotherapeutic approaches.
References
Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, Zoran MJ . Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat Rev Genet 2005; 6: 544–556.
Hirayama J, Sassone-Corsi P . Structural and functional features of transcription factors controlling the circadian clock. Curr Opin Genet Dev 2005; 15: 548–556.
Schibler U . The daily rhythms of genes, cells and organs. Biological clocks and circadian timing in cells. EMBO Rep 2005; 6 (Spec No): S9–S13.
Young ME . The circadian clock within the heart: potential influence on myocardial gene expression, metabolism, and function. Am J Physiol Heart Circ Physiol 2006; 290: H1–16.
Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM . Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol 2005; 25: 2795–2807.
Gallego M, Virshup DM . Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol 2007; 8: 139–148.
Maemura K, de la Monte SM, Chin MT, Layne MD, Hsieh CM, Yet SF, Perrella MA, Lee ME . CLIF, a novel cycle-like factor, regulates the circadian oscillation of plasminogen activator inhibitor-1 gene expression. J Biol Chem 2000; 275: 36847–36851.
Sasaki M, Yoshitane H, Du NH, Okano T, Fukada Y . Preferential inhibition of BMAL2-CLOCK activity by PER2 reemphasizes its negative role and a positive role of BMAL2 in the circadian transcription. J Biol Chem 2009; 284: 25149–25159.
Mitsui S, Yamaguchi S, Matsuo T, Ishida Y, Okamura H . Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev 2001; 15: 995–1006.
Balsalobre A, Damiola F, Schibler U . A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998; 93: 929–937.
Oishi K, Sakamoto K, Okada T, Nagase T, Ishida N . Antiphase circadian expression between BMAL1 and period homologue mRNA in the suprachiasmatic nucleus and peripheral tissues of rats. Biochem Biophys Res Commun 1998; 253: 199–203.
Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U . Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 2004; 119: 693–705.
Welsh DK, Yoo SH, Liu AC, Takahashi JS, Kay SA . Bioluminescence imaging of individual fibroblasts reveals persistent, independently phased circadian rhythms of clock gene expression. Curr Biol 2004; 14: 2289–2295.
Maemura K, Layne MD, Watanabe M, Perrell MA, Nagai R, Lee ME . Molecular mechanisms of morning onset of myocardial infarction. Ann NY Acad Sci 2001; 947: 398–402.
Nonaka H, Emoto N, Ikeda K, Fukuya H, Rohman MS, Raharjo SB, Yagita K, Okamura H, Yokoyama M . Angiotensin II induces circadian gene expression of clock genes in cultured vascular smooth muscle cells. Circulation 2001; 104: 1746–1748.
Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ . Extensive and divergent circadian gene expression in liver and heart. Nature 2002; 417: 78–83.
Maemura K, Takeda N, Nagai R . Circadian rhythms in the CNS and peripheral clock disorders: role of the biological clock in cardiovascular diseases. J Pharmacol Sci 2007; 103: 134–138.
Takeda N, Maemura K, Horie S, Oishi K, Imai Y, Harada T, Saito T, Shiga T, Amiya E, Manabe I, Ishida N, Nagai R . Thrombomodulin is a clock-controlled gene in vascular endothelial cells. J Biol Chem 2007; 282: 32561–32567.
McNamara P, Seo SB, Rudic RD, Sehgal A, Chakravarti D, FitzGerald GA . Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: a humoral mechanism to reset a peripheral clock. Cell 2001; 105: 877–889.
Chalmers JA, Martino TA, Tata N, Ralph MR, Sole MJ, Belsham DD . Vascular circadian rhythms in a mouse vascular smooth muscle cell line (Movas-1). Am J Physiol Regul Integr Comp Physiol 2008; 295: R1529–R1538.
Guney HZ, Hodoglugil U, Uluoglu C, Gorgun CZ, Ercan ZS, Abacioglu N, Zengil H . In vitro susceptibility rhythms. II. Biological-time-dependent differences in effect of beta 1- and beta 2-adrenergic agonists of rat aorta and influence of endothelium. Chronobiol Int 1998; 15: 159–172.
Otto ME, Svatikova A, Barretto RB, Santos S, Hoffmann M, Khandheria B, Somers V . Early morning attenuation of endothelial function in healthy humans. Circulation 2004; 109: 2507–2510.
Maruo T, Nakatani S, Kanzaki H, Kakuchi H, Yamagishi M, Kitakaze M, Ohe T, Miyatake K . Circadian variation of endothelial function in idiopathic dilated cardiomyopathy. Am J Cardiol 2006; 97: 699–702.
Viswambharan H, Carvas JM, Antic V, Marecic A, Jud C, Zaugg CE, Ming XF, Montani JP, Albrecht U, Yang Z . Mutation of the circadian clock gene Per2 alters vascular endothelial function. Circulation 2007; 115: 2188–2195.
Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD . Vascular disease in mice with a dysfunctional circadian clock. Circulation 2009; 119: 1510–1517.
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS . Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008; 452: 442–447.
Busik JV, Tikhonenko M, Bhatwadekar A, Opreanu M, Yakubova N, Caballero S, Player D, Nakagawa T, Afzal A, Kielczewski J, Sochacki A, Hasty S, Calzi SL, Kim S, Duclas SK, Segal MS, Guberski DL, Esselman WJ, Boulton ME, Grant MB . Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med 2009; 206: 2897–2906.
Wang CY, Wen MS, Wang HW, Hsieh IC, Li Y, Liu PY, Lin FC, Liao JK . Increased vascular senescence and impaired endothelial progenitor cell function mediated by mutation of circadian gene Per2. Circulation 2008; 118: 2166–2173.
Kunieda T, Minamino T, Katsuno T, Tateno K, Nishi J, Miyauchi H, Orimo M, Okada S, Komuro I . Cellular senescence impairs circadian expression of clock genes in vitro and in vivo. Circ Res 2006; 98: 532–539.
Hermida RC, Ayala DE, Portaluppi F . Circadian variation of blood pressure: the basis for the chronotherapy of hypertension. Adv Drug Deliv Rev 2007; 59: 904–922.
Millar-Craig MW, Bishop CN, Raftery EB . Circadian variation of blood-pressure. Lancet 1978; 1: 795–797.
Hartikainen J, Tarkiainen I, Tahvanainen K, Mantysaari M, Lansimies E, Pyorala K . Circadian variation of cardiac autonomic regulation during 24-h bed rest. Clin Physiol 1993; 13: 185–196.
Kretschmannova K, Svobodova I, Balik A, Mazna P, Zemkova H . Circadian rhythmicity in AVP secretion and GABAergic synaptic transmission in the rat suprachiasmatic nucleus. Ann NY Acad Sci 2005; 1048: 103–115.
Stern N, Sowers JR, McGinty D, Beahm E, Littner M, Catania R, Eggena P . Circadian rhythm of plasma renin activity in older normal and essential hypertensive men: relation with inactive renin, aldosterone, cortisol and REM sleep. J Hypertens 1986; 4: 543–550.
Charloux A, Gronfier C, Lonsdorfer-Wolf E, Piquard F, Brandenberger G . Aldosterone release during the sleep-wake cycle in humans. Am J Physiol 1999; 276: E43–E49.
Watanabe T, Uchiyama Y . Quantitative analyses of atrial myoendocrine cells and plasma atrial natriuretic peptides (ANP) of the rat with special reference to the twenty-four-hour variations in secretory granules and plasma ANP concentrations. Cell Tissue Res 1988; 254: 133–137.
Lemmer B, Witte K, Schanzer A, Findeisen A . Circadian rhythms in the renin-angiotensin system and adrenal steroids may contribute to the inverse blood pressure rhythm in hypertensive TGR(mREN-2)27 rats. Chronobiol Int 2000; 17: 645–658.
Scheer FA, Hilton MF, Mantzoros CS, Shea SA . Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci USA 2009; 106: 4453–4458.
Janssen BJ, Tyssen CM, Duindam H, Rietveld WJ . Suprachiasmatic lesions eliminate 24-h blood pressure variability in rats. Physiol Behav 1994; 55: 307–311.
Curtis AM, Cheng Y, Kapoor S, Reilly D, Price TS, Fitzgerald GA . Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc Natl Acad Sci USA 2007; 104: 3450–3455.
Westgate EJ, Cheng Y, Reilly DF, Price TS, Walisser JA, Bradfield CA, FitzGerald GA . Genetic components of the circadian clock regulate thrombogenesis in vivo. Circulation 2008; 117: 2087–2095.
Woon PY, Kaisaki PJ, Braganca J, Bihoreau MT, Levy JC, Farrall M, Gauguier D . Aryl hydrocarbon receptor nuclear translocator-like (BMAL1) is associated with susceptibility to hypertension and type 2 diabetes. Proc Natl Acad Sci USA 2007; 104: 14412–14417.
Wang N, Yang G, Jia Z, Zhang H, Aoyagi T, Soodvilai S, Symons JD, Schnermann JB, Gonzalez FJ, Litwin SE, Yang T . Vascular PPARgamma controls circadian variation in blood pressure and heart rate through Bmal1. Cell Metab 2008; 8: 482–491.
Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GT, Okamura H . Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med 2010; 16: 67–74.
Gumz ML, Stow LR, Lynch IJ, Greenlee MM, Rudin A, Cain BD, Weaver DR, Wingo CS . The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest 2009; 119: 2423–2434.
Colantonio D, Casale R, Abruzzo BP, Lorenzetti G, Pasqualetti P . Circadian distribution in fatal pulmonary thromboembolism. Am J Cardiol 1989; 64: 403–404.
Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T, Sobel BE, Willerson JT, Braunwald E . Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med 1985; 313: 1315–1322.
Braunwald E . Morning resistance to thrombolytic therapy. Circulation 1995; 91: 1604–1606.
Zarich S, Waxman S, Freeman RT, Mittleman M, Hegarty P, Nesto RW . Effect of autonomic nervous system dysfunction on the circadian pattern of myocardial ischemia in diabetes mellitus. J Am Coll Cardiol 1994; 24: 956–962.
Willich SN, Linderer T, Wegscheider K, Leizorovicz A, Alamercery I, Schroder R . Increased morning incidence of myocardial infarction in the ISAM Study: absence with prior beta-adrenergic blockade. ISAM Study Group. Circulation 1989; 80: 853–858.
Kobrin I, Oigman W, Kumar A, Ventura HO, Messerli FH, Frohlich ED, Dunn FG . Diurnal variation of blood pressure in elderly patients with essential hypertension. J Am Geriatr Soc 1984; 32: 896–899.
Fujita M, Franklin D . Diurnal changes in coronary blood flow in conscious dogs. Circulation 1987; 76: 488–491.
el-Tamimi H, Mansour M, Pepine CJ, Wargovich TJ, Chen H . Circadian variation in coronary tone in patients with stable angina. Protective role of the endothelium. Circulation 1995; 92: 3201–3205.
Jovicic A, Ivanisevic V, Nikolajevic R . Circadian variations of platelet aggregability and fibrinolytic activity in patients with ischemic stroke. Thromb Res 1991; 64: 487–491.
Andrews NP, Gralnick HR, Merryman P, Vail M, Quyyumi AA . Mechanisms underlying the morning increase in platelet aggregation: a flow cytometry study. J Am Coll Cardiol 1996; 28: 1789–1795.
Undar L, Turkay C, Korkmaz L . Circadian variation in circulating platelet aggregates. Ann Med 1989; 21: 429–433.
Meade TW, Mellows S, Brozovic M, Miller GJ, Chakrabarti RR, North WR, Haines AP, Stirling Y, Imeson JD, Thompson SG . Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet 1986; 2: 533–537.
Manfredini R, Boari B, Smolensky MH, Salmi R, la Cecilia O, Maria Malagoni A, Haus E, Manfredini F . Circadian variation in stroke onset: identical temporal pattern in ischemic and hemorrhagic events. Chronobiol Int 2005; 22: 417–453.
Pinotti M, Bertolucci C, Portaluppi F, Colognesi I, Frigato E, Foa A, Bernardi F . Daily and circadian rhythms of tissue factor pathway inhibitor and factor VII activity. Arterioscler Thromb Vasc Biol 2005; 25: 646–649.
Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F . Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002; 99: 3962–3970.
Madden LA, Vince RV, Sandstrom ME, Taylor L, McNaughton L, Laden G . Microparticle-associated vascular adhesion molecule-1 and tissue factor follow a circadian rhythm in healthy human subjects. Thromb Haemost 2008; 99: 909–915.
Bridges AB, McLaren M, Scott NA, Pringle TH, McNeill GP, Belch JJ . Circadian variation of tissue plasminogen activator and its inhibitor, von Willebrand factor antigen, and prostacyclin stimulating factor in men with ischaemic heart disease. Br Heart J 1993; 69: 121–124.
Kapiotis S, Jilma B, Quehenberger P, Ruzicka K, Handler S, Speiser W . Morning hypercoagulability and hypofibrinolysis. Diurnal variations in circulating activated factor VII, prothrombin fragment F1+2, and plasmin-plasmin inhibitor complex. Circulation 1997; 96: 19–21.
Rosing DR, Brakman P, Redwood DR, Goldstein RE, Beiser GD, Astrup T, Epstein SE . Blood fibrinolytic activity in man. Diurnal variation and the response to varying intensities of exercise. Circ Res 1970; 27: 171–184.
Kurnik PB . Circadian variation in the efficacy of tissue-type plasminogen activator. Circulation 1995; 91: 1341–1346.
Thogersen AM, Jansson JH, Boman K, Nilsson TK, Weinehall L, Huhtasaari F, Hallmans G . High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation 1998; 98: 2241–2247.
Andreotti F, Kluft C . Circadian variation of fibrinolytic activity in blood. Chronobiol Int 1991; 8: 336–351.
Angleton P, Chandler WL, Schmer G . Diurnal variation of tissue-type plasminogen activator and its rapid inhibitor (PAI-1). Circulation 1989; 79: 101–106.
Naito Y, Tsujino T, Kawasaki D, Okumura T, Morimoto S, Masai M, Sakoda T, Fujioka Y, Ohyanagi M, Iwasaki T . Circadian gene expression of clock genes and plasminogen activator inhibitor-1 in heart and aorta of spontaneously hypertensive and Wistar-Kyoto rats. J Hypertens 2003; 21: 1107–1115.
Schoenhard JA, Smith LH, Painter CA, Eren M, Johnson CH, Vaughan DE . Regulation of the PAI-1 promoter by circadian clock components: differential activation by BMAL1 and BMAL2. J Mol Cell Cardiol 2003; 35: 473–481.
Oishi K, Uchida D, Ohkura N, Doi R, Ishida N, Kadota K, Horie S . Ketogenic diet disrupts the circadian clock and increases hypofibrinolytic risk by inducing expression of plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol 2009; 29: 1571–1577.
Esmon CT . Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J 1995; 9: 946–955.
Sadler JE . Thrombomodulin structure and function. Thromb Haemost 1997; 78: 392–395.
Van de Wouwer M, Collen D, Conway EM . Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol 2004; 24: 1374–1383.
Young ME, Wilson CR, Razeghi P, Guthrie PH, Taegtmeyer H . Alterations of the circadian clock in the heart by streptozotocin-induced diabetes. J Mol Cell Cardiol 2002; 34: 223–231.
Young ME, Razeghi P, Taegtmeyer H . Clock genes in the heart: characterization and attenuation with hypertrophy. Circ Res 2001; 88: 1142–1150.
Kung TA, Egbejimi O, Cui J, Ha NP, Durgan DJ, Essop MF, Bray MS, Shaw CA, Hardin PE, Stanley WC, Young ME . Rapid attenuation of circadian clock gene oscillations in the rat heart following ischemia-reperfusion. J Mol Cell Cardiol 2007; 43: 744–753.
Gibson EM, Williams III WP, Kriegsfeld LJ . Aging in the circadian system: considerations for health, disease prevention and longevity. Exp Gerontol 2009; 44: 51–56.
Mohri T, Emoto N, Nonaka H, Fukuya H, Yagita K, Okamura H, Yokoyama M . Alterations of circadian expressions of clock genes in Dahl salt-sensitive rats fed a high-salt diet. Hypertension 2003; 42: 189–194.
Hurd MW, Ralph MR . The significance of circadian organization for longevity in the golden hamster. J Biol Rhythms 1998; 13: 430–436.
Penev PD, Kolker DE, Zee PC, Turek FW . Chronic circadian desynchronization decreases the survival of animals with cardiomyopathic heart disease. Am J Physiol 1998; 275: H2334–H2337.
Martino TA, Tata N, Belsham DD, Chalmers J, Straume M, Lee P, Pribiag H, Khaper N, Liu PP, Dawood F, Backx PH, Ralph MR, Sole MJ . Disturbed diurnal rhythm alters gene expression and exacerbates cardiovascular disease with rescue by resynchronization. Hypertension 2007; 49: 1104–1113.
Rudic RD, McNamara P, Reilly D, Grosser T, Curtis AM, Price TS, Panda S, Hogenesch JB, FitzGerald GA . Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation 2005; 112: 2716–2724.
Davidson AJ, London B, Block GD, Menaker M . Cardiovascular tissues contain independent circadian clocks. Clin Exp Hypertens 2005; 27: 307–311.
Tsuchiya Y, Minami I, Kadotani H, Nishida E . Resetting of peripheral circadian clock by prostaglandin E2. EMBO Rep 2005; 6: 256–261.
Yagita K, Tamanini F, van Der Horst GT, Okamura H . Molecular mechanisms of the biological clock in cultured fibroblasts. Science 2001; 292: 278–281.
Reilly DF, Curtis AM, Cheng Y, Westgate EJ, Rudic RD, Paschos G, Morris J, Ouyang M, Thomas SA, FitzGerald GA . Peripheral circadian clock rhythmicity is retained in the absence of adrenergic signaling. Arterioscler Thromb Vasc Biol 2008; 28: 121–126.
Acknowledgements
We acknowledge support for NT from the Japanese Society for the Promotion of Science and the Uehara Memorial Foundation. This study was supported by a grant-in-aid for Scientific Research from the Ministry of Education Science and Culture, Japan (21390244 to KM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takeda, N., Maemura, K. Circadian clock and vascular disease. Hypertens Res 33, 645–651 (2010). https://doi.org/10.1038/hr.2010.68
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2010.68
Keywords
This article is cited by
-
Vascular dysfunction caused by loss of Brn-3b/POU4F2 transcription factor in aortic vascular smooth muscle cells is linked to deregulation of calcium signalling pathways
Cell Death & Disease (2023)
-
Skipping breakfast during pregnancy and hypertensive disorders of pregnancy in Japanese women: the Tohoku medical megabank project birth and three-generation cohort study
Nutrition Journal (2022)
-
BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy
Protein & Cell (2020)
-
Regulation of Clock Genes by Adrenergic Receptor Signaling in Osteoblasts
Neurochemical Research (2018)
-
Intracellular high cholesterol content disorders the clock genes, apoptosis-related genes and fibrinolytic-related genes rhythmic expressions in human plaque-derived vascular smooth muscle cells
Lipids in Health and Disease (2017)
