
Abstract
Powdery mildew (PM), which is caused by the pathogen Erysiphe necator (Schw.) Burr., is the single most damaging disease of cultivated grapes (Vitis vinifera) worldwide. However, little is known about the transcriptional response of grapes to infection with PM. RNA-seq analysis was used for deep sequencing of the leaf transcriptome to study PM resistance in Chinese wild grapes (V. pseudoreticulata Baihe 35-1) to better understand the interaction between host and pathogen. Greater than 100 million (M) 90-nt cDNA reads were sequenced from a cDNA library derived from PM-infected leaves. Among the sequences obtained, 6541 genes were differentially expressed (DEG) and were annotated with Gene Ontology terms and by pathway enrichment. The significant categories that were identified included the following: defense, salicylic acid (SA) and jasmonic acid (JA) responses; systemic acquired resistance (SAR); hypersensitive response; plant–pathogen interaction; flavonoid biosynthesis; and plant hormone signal transduction. Various putative secretory proteins were identified, indicating potential defense responses to PM infection. In all, 318 putative R-genes and 183 putative secreted proteins were identified, including the defense-related R-genes BAK1, MRH1 and MLO3 and the defense-related secreted proteins GLP and PR5. The expression patterns of 16 genes were further illuminated by RT-qPCR. The present study identified several candidate genes and pathways that may contribute to PM resistance in grapes and illustrated that RNA-seq is a powerful tool for studying gene expression. The RT-qPCR results reveal that effective resistance responses of grapes to PM include enhancement of JA and SAR responses and accumulation of phytoalexins.
Similar content being viewed by others


Introduction
Powdery mildew (PM) is caused by Erysiphe necator (Schw.) Burr., a biotrophic ascomycete with a narrow host range; it is a common grapevine disease that has drawn much attention because of the economic deficits it causes. The PM fungus infects the green parts of grape plants, including leaves, stems and berries, causing decreases in yields, vine growth, winter hardiness, and fruit and wine quality. Hence, PM is an especially severe problem for grape production worldwide. Primary infection of plant tissues by conidial spores requires at least 12–15 h of continuous wetness at 10–15 °C. Under optimal temperatures, the time from infection to proliferation of new conidia can be as short as 5–6 days. Host responses to PM have been characterized in various resistant species, such as Vitis labrusca, Vitis rupestris1 and Vitis aestivalis.2 A previously determined resistance mechanism consists of physical barriers that obstruct penetration of the host epidermis by the developing germ tubes of germinated PM conidia. A previous study has shown that the R-gene Resistance against U. necator 1 (Run1), which is mediated through a hypersensitive response, is responsible for PM resistance in Muscadinia rotundifolia.3 Using the Vitis GeneChip, Fung et al.4 reported that 625 transcripts in V. vinifera and only three transcripts in V. aestivalis were involved in host responses to PM. Fekete et al.5 identified 25 PM-responsive genes in V. vinifera (cv. Cabernet Sauvignon) that were upregulated specifically in response to E. necator. These previous studies provide valuable information regarding PM interactions in European and North American grape species.
Wine grape varieties vary in their susceptibility to PM because they are derived from Vitis vinifera L. (Vv), a species that was not exposed to this pathogen during its evolution.6 In contrast, the Chinese wild Vitis species co-evolved with E. necator and possess various levels of PM resistance, making these species a valuable resource for studying PM resistance in grapes. Among wild Vitis species, the Chinese wild Vitis pseudoreticulata (Vp) has been shown to possess potentially durable, non-race-specific PM resistance.7,8 Disease-resistance genes have successfully been transferred from wild species of Chinese Vitis to V. vinifera. Thus, understanding the genetic and molecular basis of disease resistance and identifying the key genes responsible for resistance in resistant germplasm should provide us with valuable knowledge regarding the use of molecular breeding to develop new grape varieties that exhibit PM resistance while preserving high fruit quality.
Mechanisms of PM resistance in the Chinese wild species of V. pseudoreticulata and Vitis quinquangularis were investigated using a suppression subtractive hybridization cDNA library that was constructed from E. necator-infected leaves.8 Xu et al.9 constructed a cDNA library from PM-inoculated Vitis pseudoreticulata and identified the expressions of five PM-defense-related sequence tags. Research efforts focused on gene expression profiling in V. pseudoreticulata resistance have demonstrated the expression of pathogenesis-related (PR) genes, such as PR10, a transcription factor that may contribute to PM resistance in grapevines.10 Others genes may also play important roles in PM resistance; these include VpWRKY1,2,11 ethylene response factor,12 VpWRKY3,13 VpNAC1,14 ring finger protein VpRFP1 and its promoter,15,16 the heat shock transcription factor VpHsf1, retinoblastoma-related,17 stilbene synthase (STS),18 glyoxal oxidase19 and aldehyde dehydrogenases.20 Zhang et al.21 used the Vitis microarray to analyze gene expression in V. pseudoreticulata and detected expression of 11 906 of the 16 602 genes known to exist on the microarray.21
To identify potential resistance mechanisms and resistance-related genes, the present study applied RNA-seq to obtain a more accurate and comprehensive profile of global gene expression patterns in V. pseudoreticulata in response to POM infection. The study used Baihe 35-1, a clonal selection of Chinese wild V. pseudoreticulata that exhibits high levels of resistance to PM. Comparison of two libraries constructed from leaf tissues that were collected prior to and after PM inoculation demonstrated that PM inoculation results in strong ETI, basal defense and systemic acquired resistance (SAR) responses and facilitates secondary metabolism and biosynthesis of lignin phytoalexins and anthocyanin. Analyses of secretory proteins revealed that grape cells may secrete PR proteins, as well as lectin, germin-like protein, glycosyl-hydrolase-family protein that has chitinase insertion domain, protease and glucosidase. Baihe 35-1 cells also release a pectate lyase that may strengthen resistance in the surrounding cells via pectate-activating wall-associated plant receptor protein kinases (RPKs) (e.g., WAK1).
Results
Read sequencing, quantification and assembly
A total of 5.28 Gb and 5.02 Gb 90-base reads were sequenced from libraries constructed from PM-infected (BH-T) leaf tissues from one to seven days each 24 h and from non-vaccinated (BH-C) leaf tissues as a control, respectively (Table 1). After removing adaptor sequences and discarding low-quality reads, (i.e., those containing more than 5% unknown bases and/or over 50% bases with a quality value ≤5), 51 493 326 and 50 507 790 clean reads were obtained from the BH-C and BH-T libraries, respectively. The raw sequencing data have been uploaded to the NCBI Sequence Read Archive; the BH-C accession is SRR1522047, and the BH-T accession is SRR1535993.
The reads were compared with the V. vinifera grape reference genome (http://www.genoscope.cns.fr/) using SOAPaligner/soap2. Only reads with less than five base-pair mismatches were counted and used in subsequent analyses. The data in Table 1 show that a substantial proportion of reads (76.56% in the BH-C library and 76.97% in the BH-T library) were able to be aligned with the ‘Pinot Noir’ genome, whereas 26 055 438 (50.60% of reads) and 25 429 109 (50.35% of reads) in the BH-C and BH-T libraries, respectively, aligned with predicted ‘Pinot Noir’ genes. Furthermore, 26 536 965 (51.53%) reads in the BH-C library and 25 919 400 (51.32%) in the BH-T library were matched to a single gene sequence in the ‘Pinot Noir’ genome (Table 1).
Comparison of gene expression in BH-C and BH-T samples
RPKM (Reads Per Kilobase per Million mapped reads) were used to estimate gene expression levels in both libraries. Transcripts detected with a log2 Ratio (BH-T/BH-C) ≥1 (indicating a two-fold difference) were considered to be differentially expressed. The results indicated that a total of 21 609 genes met these criteria (Figure 1). Transcripts with twofold greater abundance (2073 red dots) and two-fold lower abundance (4468 green dots) represent upregulated and downregulated transcripts, respectively, in response to PM infection. The Venn diagrams in Figure 2 summarize the information obtained from the two libraries. The Venn diagram on the right indicates that 755 genes were uniquely expressed in the BH-C (control) library, while 1145 genes were uniquely expressed in the BH-T (PM-infected) library; 19 708 genes were expressed in both libraries. The Venn diagram on the right indicates that, among the 6541 differentially expressed (DEGs), 281 genes were uniquely upregulated in the BH-C library, 121 were uniquely downregulated in the BH-T library, and 6139 were either up- or downregulated in the DEGs common to both libraries. Annotations of assembled DEG sequences were conducted via sequence similarity searching against the NR protein database (non-redundant protein sequence database, NCBI) using BLASTx. The E-value cutoff was set as 1×10−6. Out of a total of 6541 DEG sequences, 6508 could be identified as grape genes and annotated (Supplementary Table S1).

Distribution of gene expression levels. Red dots (2073) represent more prevalent transcripts in the infected PM leaf library; lower frequencies in the infected tissue are represented by green dots (4468); and transcripts showing no significant differences are represented by blue dots (15 068).

Venn diagrams of gene expression in control (BH-C) and PM-inoculated (BH-T) samples. The Venn diagram on the left indicates that a total of 21 609 sequences were identified in both libraries (combined) and that 755 were unique to the BH-C library, 1145 were unique to the BH-T library and 19 708 were common to both libraries. The Venn diagram on the right indicates that 281 DEGs were uniquely upregulated in the BH-C library, 121 were downregulated in the BH-T library and 6139 were either up- or downregulated in one of the libraries.
Functional annotation and classification
The differentially expressed genes associated with responses to PM infection were categorized according to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses.
GO annotation
BLASTx (E-value <10−6) of DEGs was conducted against the GO sequence database (http://www.geneontology.org). GO annotations could be assigned to a total of 6067 genes; 3599 of these were grape genes, and another 2468 were Arabidopsis genes. In all, 2145 GO terms were assigned to the 6067 GO-annotated genes. Collectively, 4735 DEGs were classified in the ‘biological process’ category; 4370 DEGs were classified in the ‘molecular function’ category; and 5586 DEGs were classified in the ‘cellular component’ subcategories (Table 2). The top 20 (i.e., categories with the highest number of gene annotations) GO terms indicated PM-induced defense and immune responses in grapes and stimulated secondary metabolism at the mean time. The GO analysis corresponded well with expression levels of defense-related genes. Up- and downregulated genes and their associated GO terms revealed more information about the resistance response in grapes to PM (Table 2). Defense and immune response systems were stimulated in PM-infected grapes, and secondary metabolism was also enhanced, as these are processes associated with systemic acquired resistance. Together, these processes form the basis of PM resistance in V. pseudoreticulata, Baihe 35-1. In addition to these defense responses, the host response to PM also involved decreased methylation and cell proliferation metabolism.
The top 20 GO terms associated with the upregulation of genes in response to PM indicate three main ways in which grapes respond to PM. The major defense response includes an ‘immune response’ and ‘systemic acquired resistance’. Grape cells infected by PM perceive the pathogen, express a recognition signal, and subsequently initiate a regulated defense response. The data in Table 3 indicate that salicylic acid (SA) and jasmonic acid (JA) constitute the main hormone signal transduction and response pathways that are induced in grapes by PM. It appears that activating these two hormone signaling and transduction pathways stimulates the SAR program. The second major defense-related response is associated with the stimulation of secondary metabolism and small molecule biosynthesis. ‘Phenylpropanoid biosynthesis’ is a major secondary metabolic process and plays an important role in plant defense responses. It is involved in the biosynthesis of SA, lignin and anthocyanin. The stimulation of ‘small molecule biosynthesis’ in response to PM infection could be associated with the biosynthesis of small-molecule hormones, such as SA, ET, JA and ABA, each of which plays a role in plant immunity. The last defense response to PM in grapes is more ‘offensive’ and occurs via the stimulation of ‘transport’ and secretion (‘localization’) GO categories, an action that may be aroused by the secretion of PR proteins. The downregulation of genes annotated with GO terms related to cell replication and methylation may enable cells to allocate more energy to the expression of defense-related genes and processes.
Pathway annotation
To explore the biochemical pathways in which DEGs were involved, a pathway analysis utilizing the KEGG was conducted with an E-value cutoff of <0.05. Eight significantly enriched pathways associated with the DEGs were identified (Table 4). The ‘Ribosome’ pathway was related to the biogenesis of proteins, DNA and RNA. Five pathways were related to metabolism, such as ‘flavonoid biosynthesis’. ‘Plant–pathogen interaction’ and ‘plant hormone signal transduction’ pathways were also associated with PM-related gene expression.
In the plant–pathogen interaction pathway, three grape membrane receptor genes, WAK1, WAK10 and BAK1, were upregulated (Figure 3). These membrane receptors regulate the SA defense response via a MAPKKK cascade. Calcium-mediated signaling was also activated, as the ROS pathway was responsible for the hypersensitive response. Avirulence (AVR) is also a signal that leads to a strong defense response. In plant hormone signal transduction pathways, although the GO analysis indicated stimulation of SA and JA responses, genes associated with the SA response, such as WRKY genes, did not exhibit differential expression. This was also true for JA response genes. Secondary metabolism, such as flavonoid biosynthesis, was also stimulated in response to PM. In this regard, genes responsible for the biosynthesis of lignin, phytoalexins and anthocyanin were upregulated (Figure 4).

PAMPs activate PRRs. Boxes indicate the critical enzymes comprising the pathway. Enzymes colored in red or green indicate upregulated or downregulated genes, respectively. Genes in black are those with no differences in expression, whereas enzymes shown in black are those that were not identified in the present study. AVR, avirulence protein; BAK1, brassinosteroid insensitive 1-associated receptor kinase 1; CNGC, cyclic nucleotide gated calcium channel; OG, oligogalacturonides; HR, hypersensitive response; PAMP, pathogen-associated molecular pattern; PRR, pattern-recognition receptor; RBOH, respiratory burst oxidase homolog; ROS, reactive oxygen species; SA, salicylic acid; WAK1, wall-associated RPK.

Flavonoid biosynthesis pathway. Boxes indicate the critical enzymes comprising the pathway. Enzymes colored in red or green indicate upregulated or downregulated genes, respectively. Enzymes in black are those with no differences in expression, whereas enzymes shown in black are those that were not identified in the present study. AAO, aldehyde oxidase; ANS, anthocyanidin synthase; BA2H, benzoic acid 2-hydroxylase; BZL, benzoyl-CoA ligase; CAD, cinnamyl alcohol dehydrogenase; CCoAOMT, caffeoyl-CoA O-methyltransferase; CCR, cinnamoyl CoA reductase; C3H, p-coumarate 3 hydroxylase; C4H, cinnamate 4-hydroxylase; CHI, chalcone isomerase; CHS, chalcone synthase; 4CL, 4-coumarate: CoA ligase; COMT, caffeic acid o-methyltransferase; DFR, dihydroflavonol-4-reductase; F3H, flavanone 3-hydroxylase; F5H, ferulate 5-hydroxylate; FLS, flavonol synthase; HCT, hydroxycinnamoyl transferase; ICS, isochorismate synthase; IPL, isochorismate pyruvate lyase; PAL, phenylalanine ammonia-lyase.
R gene and Transcription factor analysis
All of the DEGs were queried against the Plant Resistance Gene database (http://prgdb.crg.eu/)22 using BLASTp. A total of 318 putative R genes were identified, 132 of which were upregulated and 186 of which were downregulated in the BH-T transcriptome. A list of the identified R-genes is provided in Supplementary Table 1. The upregulated genes belonged mainly to the NL, receptor-like protein (RLP), CNL and N classes of R genes. The Mlo-like R-gene class constitutes the majority of upregulated genes (Table 5).
The DEGs were also queried against the PlnTFDB (http://plntfdb.bio.uni-potsdam.de/) database using BLASTx to identify putative transcription factors. Among the DEGs, 642 putative transcription factors (160 upregulated and 481 downregulated) were identified (Figure 5). The largest transcription gene family present in the DEGs was the MYB family, followed by the basic helix–loop–helix family, the ethylene-responsive element binding factor family, the homeodomain–leucine zipper family and the C2H2 family, respectively.

Distribution of up- and downregulated transcription factors in gene families. The distribution of up- and downregulated transcription-factor gene-family members.
Secretory protein prediction
Secreted proteins (SPs) bear a short stretch of amino acids (signal sequence) at their N-terminus that targets the protein for secretion. The SPs identified among the DEGs were analyzed using SignalP software (http://www.cbs.dtu.dk/services/SignalP), which identifies signal sequences and their cleavage sites. Of the 183 DEGs identified as putative secretory proteins (Supplementary Table S3), 60 upregulated SPs appear to contain signal sequences with mean S scores >0.48 (part of upregulated SP genes are shown in Table 6).
RT-qPCR analysis
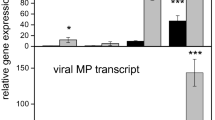
To further analyze the response of V. pseudoreticulata to PM and to verify the results obtained by RNA-seq, the expression levels of 16 defense-related genes were analyzed by RT-qPCR. The expression levels of the selected genes were measured at 12, 24, 36, 48, 72, 96 and 120 h post inoculation (hpi) with PM. Leaves from un-inoculated plants served as the control (Time 0). Results are presented in Figure 6. The four SA pathway genes, ICS1 (isochorismate synthase 1), SAMT (SA carboxyl methyltransferase), NPR1 (non-expressor of pathogenesis-related genes 1) and PR1, contribute to SA biosynthesis, methylation, signal transduction and response, respectively. The expression trend exhibited by these four genes indicates that the SA pathway is downregulated in the first 36 hpi but upregulated afterwards (Figure 7). A similar pattern was observed for genes related to anthocyanin biosynthesis. In contrast, genes related to JA biosynthesis, such as AOS (allene oxide synthase) and PDF (plant defensin), as well as genes related to SAR, such as DIR (lipid-transfer protein), exhibited inverse patterns. These inverse expression patterns were also exhibited by phytoalexin genes. The RbOH gene was downregulated in the first 24 hpi, upregulated in the 24–72 hpi, and then downregulated after 72 hpi. Importantly, regarding the flavonoid biosynthesis pathway, PAL (phenylalanine ammonia lyase) and STS are upregulated during the first 72 hpi, while CCOOMT (S-adenosyl-l-methionine:trans-caffeoyl-CoA 3-O-methytransferase), CHS (chalcone synthase) and ANS (leucoanthocyanidin dioxygenase) are downregulated. These data suggest that phytoalexin accumulation, rather than SA, anthocyanin, or lignin biosynthesis, is the main response of V. pseudoreticulata to PM in the first 72 hpi.

RT-qPCR analysis of sixteen selected DEGs. RT-qPCR analyses of the expression of sixteen different DEGs in response to inoculation with PM. Samples were collected at 12, 24, 36, 48, 72, 96 and 120 hpi. Uninoculated leaves served as the control. All data were normalized to the expression level of actin. Data represent the fold change of RQ (relative quantification) at each time point in infected vs. control samples (Time 0). Bars represent standard deviations calculated from three biological replicates.

Up- or downregulation of different pathways in response to PM inoculation. The green bar indicates that the pathway was downregulated, and the red bar indicates that the pathway was upregulated.
Discussion
A high-throughput DNA sequencing technology, RNA-seq, was used to study the resistance response of V. pseudoreticulata to PM. Strict filtering and conservative matching of the sequencing data to the grape reference genome were used in the expression profiling analysis. The obtained reads were assembled prior to mapping to obtain longer sequence reads (contigs), which facilitated mapping and annotation. In total, more than 50% of the sequenced reads were assigned to genes and used for gene expression profiling. This proportion was relatively low compared to other transcriptome studies utilizing RNA-seq, an observation that may be explained, in part, by the fact that most of the sequences obtained represented sequences from Erysiphe necator, the causal agent of PM. Additionally, grapes have a large number of unknown genes and expressed elements. The remain sequenced reads were occurred among currently unknown transcripts (∼10%, reads in intergenic regions) or splice variants, novel exons or untranslated regions (∼16%) and RNA genes. Therefore, the result of 50% is reasonable (Supplementary Fig. S1).
GO and pathway enrichment (KEGG) analyses of DEGs were conducted after the DEGs had been annotated by BLASTx against the NR database using a cutoff E-value <10−6. Because many grape genes are still unknown, much less functionally annotated, 6109 of the DEGs were assigned GO terms based on their homology to Arabidopsis thaliana genes (http://arabidopsis.org, TAIR10) using BLASTp with a cutoff E-value <10−6. The E-value distribution of DEGs that can be matched to A. thaliana genes is shown in Supplementary Fig. S2b. A GO enrichment analysis provided information related to the function of the DEGs that respond to PM.
Pathway enrichment analysis (KEGG) of the DEGs revealed that the pathways were affected by PM. It is not surprising that the majority of DEGs were categorized in the GO term ‘ribosome’ pathway. This suggests that the resistant grape genotype, Baihe-35-1, utilizes new ribosomes or changes ribosome components to facilitate more rapid syntheses of additional proteins, such as PR proteins. At least two responses were screened out of the other affected pathways in which the resistant Baihe-35-1 responds to PM. The first response is reflected in the obtained GO terms ‘plant–pathogen interaction’ and ‘plant hormone signal transduction’, suggesting that Baihe-35-1 perceives pathogen invasion, transmits a signal and induces a resistance response. The second major response is reflected in the obtained KEGG pathway, ‘flavonoid biosynthesis’.
Induced defense responses
In plants, innate immunity is triggered through the response of pattern recognition receptors to pathogen-associated molecular patterns. This provides the first line of inducible pathogen-associated molecular pattern-triggered immunity.23,24 Three RPKs, WAK1, WAK10 and BAK1, exhibited increased expression levels in response to PM inoculation. Wall-associated RPKs (WAK1 and 10) function to prolong or amplify immune responses, bind pectin and oligogalacturonides and modulate both immunity and development as secondary plant signals.25,26 BAK1 does not have a direct influence on elicitor perception, but it can rapidly form a complex with FLS2 after elicitation.27 BAK1 is a central regulator of plant immunity and, consequently, the target of several pathogen virulence effector molecules.28 The two RPKs, WAK1 and 10, transduce a signal via three MAPK cascades, specifically the partially redundant MPK3/6 and MPK4 (Figure 2), which are associated with innate immunity. These cascades lead to the activation of the transcription factors WRKY22 and WRKY33, which regulate the defense response.29 A study of the response of Arabidopsis to Botrytis cinerea infection revealed that the loss of WRKY33 function results in inappropriate activation of the SA-related host response (like PR1) and the downregulation of JA-associated responses.30 Defense responses are also initiated when R proteins in the host cells detect effector/AVR products of the pathogen.31 In barley, intracellular mildew A R proteins function in the nucleus to confer resistance against PM.31 Recognition of the fungal avirulent A10 effector by intracellular mildew A10 induces nuclear associations between a receptor and WRKY40 transcription factors.32 Upregulation of MPK3, WRKY33 and WRKY40 were also induced by PM in PM-susceptible Vitis vinifera ‘Cabernet sauvignon’, but no discrepancies were observed in PM-resistant Vitis aestivalis ‘Norton’.4 BAK1 and WAK1 are correlated with the transduction of a calcium signal via ROS, which gives rise to a hypersensitive defense response to PM.33,34
Small-molecule hormones fine-tune the defense response
Phytohormones, such as ABA, JA, ET, SA, auxin, GA, cytokinin and brassinosteroids, play key roles in the complex signaling cascades that form current models of defense responses.24 Two responses of the hormones SA and JA are indicated by the FDR <10−5 in the GO analysis. This conclusion is supported by the identification of SA and JA signaling pathways, biosynthesis and metabolic GO terms in response to PM. The identification of the GO terms ‘systemic acquired resistance, and salicylic acid-mediated signaling pathway’ suggest that SA signaling plays a strong active role in the response of grapes to PM.
Plant resistance to biotrophic pathogens is classically thought to be mediated through SA signaling.24 Pathogen induction of SA, which is synthesized from chorismate by isochorismate synthase (ICS1),35 increases the SA defense response. Downregulation of the SA biosynthesis gene, AAO (aldehyde oxidase), and upregulation of SAMT, SOT12 (salicyloyl-L-aspartate (SA-Asp), sulphotransferase 12), MES2 (methyl esterase 2), and SGT1 (salicylic acid glucosyltransferase 1) to modify SA,36 strongly suggests that Vitis pseudoreticulata accumulates a high concentration of SA after 36 hpi.4 Further analysis of the SA signaling pathway and SA response indicated that activation of the SA signaling pathway is not sustained during the 7 days after inoculation monitored in this study. This was evidenced by the flat response of NPR1 and SA-response WRKY genes.29 PR1 expression was downregulated in the first 24 hpi, as PR1 expression in PM-resistant V. aestivalis response to PM,4 supporting the SA pathway response. It is possible that inhibition of SA resulted from an adjustment in the SA signaling pathway and the associated SA response by JA, ET, ABA, auxin and gibberellin signaling or by the presence of PM secreted proteins. These data may indicate that the SA pathway is regulated by MAP4 or pathogen elicitors.24
Jasmonates are generally associated with defense responses against herbivores and necrotrophic pathogens and, more recently, have been implicated in resistance to biotrophs, such as powdery and downy mildews in grapevines. The binding of WRKY40 to the JAZ8 (jasmonate ZIM-domain) promoter facilitates powdery mildew infection, as confirmed by chromatin immunoprecipitation assays. Chromatin immunoprecipitation assay qPCR experiments also suggested the direct negative regulation of JAZ1 and JAZ5 by WRKY33 upon infection.31 This is also in agreement with RNA-seq data obtained in this study. Recent studies have reported that lipid signals, such as JA-derived molecules, are required for SAR in Arabidopsis.37 The elicitors MeJA, JA, cyclodextrins and Na-orthovanadate, when used individually or in combination on grape plant cell cultures, elevate transcript levels of genes coding for pathogenesis-related proteins (acidic class IV chitinase, serine protease inhibitor, polygalacturonase-inhibiting protein and β-1,3-glucanase) and induce or enhance the production of stilbene phytoalexins.38 In addition, a study of the role of various partners involved in MeJA defense responses reported that an influx of calcium through the plasma membrane appears to be essential for MeJA-induced stilbene accumulation.39,40 MeJA-elicited responses that are mediated by ROS, in which superoxide anions play a greater role than H2O2, were also observed.
Secondary metabolites in the defense response
Secondary metabolites, including terpenoids, phenylpropanoids and nitrogen-containing substances, significantly impact a plant's ability to protect itself from invading pathogens.41 In the present study, GO and pathway analyses identified the contribution of secondary metabolites, via flavonoid biosynthesis, to the defense of V. pseudoreticulata against PM. Analysis of this pathway in our study revealed a wide range of genes involved in multiple branches of the phenylpropanoid pathway.41 Similar results have been reported in Arabidopsis, rice, tomato and legume plants, such as Medicago truncatula, alfalfa, pea and soybean.38 Other reports have also provided evidence that lignin, flavonoid phytoalexins and phenolic compounds play important roles in the defense response of grapes to PM.38 The rapid production of resveratrol and its transformation into viniferins appear to enhance powdery mildew resistance in grapevine cultivars.42 Trans-ε-viniferin and trans-δ-viniferin accumulate in 48–72 hpi in the highly PM-resistant grape Vitis candicans.42 The sustainable growth of STS expression in V. pseudoreticulata in the first 72 hpi has been found to differ significantly from that of V. aestivalis and V. vinifera, which only exhibited upregulation at 12 hpi and 48 hpi, respectively.4 Our results indicate that phytoalexin biosynthesis also plays a major role in the resistance response mediated by secondary metabolism in the first 72 hpi. Although phytoalexin biosynthesis-related pathways, the stilbene pathway, systemic acquired resistance, cell wall metabolism and phenylpropanoid metabolism, are represented by DEGs,43 our study is the first to provide information pertaining to the timing of induction of different branches of the phenylpropanoid biosynthesis pathway using RNA-seq and RT-qPCR. The ability of PM to infect V. pseudoreticulata decreases after 36 hpi. The strengthened SA response, reduced JA response and thickening of cell walls may facilitate V. pseudoreticulata defense against PM from 72 hpi to 120 hpi.
Secretory protein
Investigation of the secretome of Magnaporthe oryzae under nitrogen starvation conditions, which mimics fungal infection, revealed that cell-wall hydrolase enzymes, protein and lipid hydrolases, and reactive oxygen species-detoxifying proteins were the main contributors to resistance.44 Further research demonstrated that upregulation of glycosyl hydrolase-encoding transcripts occurred in rice in response to the blast fungus (Magnaporthe oryzae), suggesting that enzymes involved in cell wall degradation play an important role in plant defense and pathogen attack.45 In our study, four glycosyl hydrolases (GSVIVT01003718001, GSVIVT01009700001, GSVIVT01007898001, GSVIVT01001064001), six lipases (GSVIVT01009957001, GSVIVT01000777001, GSVIVT01009315001, GSVIVT01009959001, GSVIVT01008272001, GSVIVT01007995001), two PR 5 thaumatin-like proteins (GSVIVT01008918001, GSVIVT01009646001), a RING finger protein (GSVIVT01009098001), a germin-like protein (GSVIVT01000071001), two proteinases (GSVIVT01006968001, GSVIVT01009810001) and a nodulin (GSVIVT01001982001), were upregulated in response to PM inoculation. PR-5s,46 RING finger protein15 and germin-like protein (GLP)47 are known to participate in PM resistance. A study on a segregation of grape populations derived from a ‘Merzling’ × ‘Teroldego’ cross-exhibited significant transcriptional activation of PR2 and PR5 genes, which have been shown to be activated by ethylene,46 in genotypes that are resistant to Plasmopara viticola. Further research confirmed that the grapevine PR5 protein, VvTLP-1, significantly inhibits in vitro spore germination and hyphal growth of Elsinoë ampelina, the causal agent of grape anthracnose.48 Two other grape PR5s exhibited strong antifungal activity in vitro, which inhibited the growth of Phomopsis viticola and Botrytis cinerea mycelia.49 In the present study, the expression of PR5, STS and Rboh, as determined by RT-PCR, showed an interesting expression pattern (Figure 6). PR5 and STS transcripts were elevated in the first 36 hpi, which could have initiated and accelerated hypersensitive cell death via a second oxidative burst.50 Paradoxically, Rboh, a primary signal that is generated by microtubule disruption and that activates STS gene expression, was downregulated in the first 36 hpi. These data suggest that the STS gene was activated by the MAPK-signaling pathway rather than by Rboh.51 GLPs are also expressed by plants in response to invasion by fungal pathogens, bacteria, viruses and Erysiphe nectar.38 These genes have reportedly been induced in response to powdery mildew (Blumeria graminis f. sp) infection in barley (HvGLP4) and wheat (TaGLP4).52,53 Godfrey et al. reported that VvGLP3 was induced in ontogenically resistant, but not ontogenically susceptible, grapevine berries following inoculation with PM.47 In our study, GLP was upregulated within 12 hpi, and its expression peaked at 72 hpi, suggesting that GLP plays an important role in the defense response over a long period of time. Perhaps of greater interest, SP (GSVIVT01008053001), a pectate lyase, was upregulated, perhaps to increase the level of pectate in the cell wall, thereby providing greater opportunity for WAK1 recognition to enhance and transmit a defense signal indicating that PM is near or is trying to invade the host plant cell.
R-gene expression
There is strong selection pressure for the evolution of new plant disease-resistance genes (R-genes), which play key roles in recognizing proteins expressed by specific pathogen-derived AVR genes and provide stable resistance for many generations.54 The CNL class of resistance genes is comprised of genes encoding proteins with a coiled-coil domain, a nucleotide binding site and a leucine-rich repeat (CC-NB-LRR). NB-containing protein fragments of RPS2 require the CC domain to initiate ectopic cell death in Arabidopsis.55 Among the DEGs in the present study, 26 upregulated CNL R-genes were identified, two of which are RPS2 (GSVIVT01021921001, GSVIVT01037631001). The RLP class is characterized by proteins with a serine–threonine kinase-like receptor domain and an extracellular leucine-rich repeat (ser/thr-LRR). Twenty-three upregulated RLP R-genes were among the identified DEGs, including one BAK1 (GSVIVT01029816001) and two MRH1 (GSVIVT01032772001, GSVIVT01021228001) genes. The MLO-like class (Mildew Locus O, MLO) of R-genes is required for successful host-cell invasion by powdery mildew fungi. VvMLO3 was significantly induced in grape leaves within 8 hpi and coincided with the commencement of fungal penetration. VvMLO3 clustered within the same clade as Arabidopsis and tomato MLO genes that have been implicated in PM susceptibility.38 Four MLO-like R-genes were upregulated in V. pseudoreticulata in the present study. The elevated expression of two VvMLO3 (GSVIVT01025652001, GSVIVT01025160001) genes suggest that V. pseudoreticulata resistance to PM is not based on preventing host-cell penetration by the fungus.
Conclusions
RNA-seq was utilized to reveal the transcriptomic response of resistant V. pseudoreticulata Baihe 35-1 to inoculation with E. necator, the causal agent of powdery mildew. This study provided a comprehensive analysis of DEGs that responded to PM inoculation. We identified 2073 upregulated and 4468 downregulated genes. Further analysis of the DEGs, using a time course of 0, 12, 24, 36, 48, 72, 96 and 120 hpi for the expression of selected genes by RT-qPCR, revealed contrasting expression patterns for genes associated with JA (upregulated) and SA (downregulated) during the first 36 hpi (Figure 7). These data suggest that the JA pathway, SAR, accumulation of phytoalexins and ROS-dependent hypersensitive response play key roles in V. pseudoreticulata resistance to PM. Analysis of the predicted secretory proteins identified several interesting proteins that may affect communication between the host cell and the pathogen. The role of these secretory proteins in V. pseudoreticulata resistance to PM warrants further investigation.
Methods
Plant materials and pathogen infection
Fourteen young plants of the clonal V. pseudoreticulata, accession ‘Baihe-35-1’, were grown in 10-cm pots that were filled with a mixture of 60% vermiculite and 40% meadow soil and cultured in growth chambers employing 16 h light/8 h dark cycles at 25–26 °C. E. necator-infected leaves were collected from field-grown V. vinifera. Inoculation by E. necator was performed on Chinese wild V. pseudoreticulata ‘Baihe-35-1’ under field conditions as previously described.56 Three leaves from each V. pseudoreticulata ‘Baihe-35-1’, were inoculated with E. necator under field conditions, as described previously,36 and samples were collected every 24 h for 7 days. Control samples were harvested from water-treated leaves under the same conditions. The inoculated leaves were immediately covered with paper bags to prevent infection by other pathogens, were frozen immediately in liquid nitrogen upon collection and were stored at −80 °C until further use.
Illumina sequencing
Samples, consisting of inoculated leaves and water-treated leaves collected from 1 to 7 d, were pooled for RNA isolation and library construction. Comparable control leaves were treated identically in parallel. Total RNA was isolated from the collected leaves using a modified guanidine thiocyanate method described by Zhang et al.57 Fragmentation buffer was added to fragment mRNA into short fragments. These short fragments were used as templates, with random hexamer-primers used to synthesize the first-strand cDNA. The second-strand cDNA was synthesized using buffer, dNTPs, RNase H and DNA polymerase I. Short fragments were purified using a QiaQuick PCR extraction kit and resolved with EB buffer for end repair then adding poly(A). The short fragments were then connected with sequencing adaptors. For amplification with PCR, we selected suitable fragments as templates, based on the results of agarose gel electrophoresis. A Solexa HiSeqTM2000 sequencer was employed to sequence the constructed libraries (BGI, Shenzhen, China). The sequenced cDNA fragments were approximately 90 bp in length. The raw sequencing data have been submitted to NCBI Sequence Read Archive.
Sequence annotation and assessment of gene expression
The raw reads obtained were preprocessed by removing the adaptor sequences and discarding empty or low-quality sequences, and the reads were mapped to the grape (V. vinifera cv. Pinot Noir) reference genome (http://www.genoscope.cns.fr/) using SOAPaligner/soap2 with default parameters. Only a complete match or those with <5 base-pair mismatches were used in further analyses. RPKM values for gene sequences in the BH-T and BH-C libraries were used to estimate gene expression levels and determine differential expressions of genes that responded to PM infection using a cutoff value of |log2 ratio<BH-T/BH-C>|≥1.
GO and pathway enrichment analysis
DEGs were annotated using blastx against the NCBI NR database using a cutoff E-value of <10−6. The functions of DEGs were characterized using GO terms. Using the agriGO database,58 3599 grape sequences were associated with GO terms. The remaining 2468 unannotated DEGs were characterized by GO terms based on the homology of grape DEGs with Arabidopsis thaliana genes. The second method used for DEG characterization was based on KEGG pathway enrichment analysis. Pathways with Q values <0.05 are significantly enriched in DEGs. The Q value was used to determine the p-value threshold.59
Transcription factor and R-gene analysis
A BLASTx against the PlnTFDB (http://plntfdb.bio.uni-potsdam.de/), using a cutoff E-value of <10−6, was employed to identify DEGs that were putative transcription factors. DEGs were also queried against the Plant Resistance Gene database (http://prgdb.crg.eu/) using BLASTp.
Secretory protein prediction
SignalP4.1 (http://www.cbs.dtu.dk/services/SignalP) was employed, using default parameters, to predict which DEGs coded for secretory proteins.60
RT-qPCR
Sixteen genes were selected among the DEGs that responded to PM infection and were subsequently subjected to a time-course analysis of gene expression in response to PM using RT-qPCR. A list of the gene-specific primers designed against the 3′ untranslated region of each selected gene is provided in Supplementary Table S2. Samples were prepared using the same method mentioned above, and total RNA was isolated from leaves at 0, 12, 24, 36, 48, 72, 96 and 120 hpi using a Promega EZ RNA KitTM. cDNA libraries were synthesized using a TaKaRa:PrimeScriptTM II 1st Strand cDNA Synthesis Kit. A melting-curve analysis of the amplification products was performed to determine the specificity of the primers. The data analysis used IQ-5 by .
Authors' contributions
YX and YJW conceived and designed the experiments. ZQL and RQL performed the experiments. KW and YX analyzed the data. KW ZQL and RQL contributed reagents/materials/analysis tools. KW and YX wrote the manuscript.
References
Doster M, Schnathorst W . Comparative susceptibility of various grapevine cultivars to the powdery mildew fungus Uncinula necator. Am J Enol Viticulture 1985; 36: 101–104.
Giannakis C, Bucheli C, Skene K, Robinson S, Scott NS . Chitinase and β-1,3-glucanase in grapevine leaves: a possible defence against powdery mildew infection. Aust J Grape Wine Res 1998; 4: 14–22.
Donald T, Pellerone F, Adam-Blondon AF, Bouquet A, Thomas M, Dry I . Identification of resistance gene analogs linked to a powdery mildew resistance locus in grapevine. Theor Appl Genet 2002; 104: 610–618.
Fung RW, Gonzalo M, Fekete C et al. Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol 2008; 146: 236–249.
Fekete C, Fung RW, Szabó Z et al. Up-regulated transcripts in a compatible powdery mildew–grapevine interaction. Plant Physiol Biochem 2009; 47: 732–738.
Mullins MG, Bouquet A, Williams LE . Biology of the Grapevine. Cambridge: Cambridge University Press, 1992.
Wang ZY . Evaluation of foliar resistance to Uncinula necator in Chinese wild Vitis species. Vitis 1995; 34: 159–164.
Wan Y, Schwaninger H, He P, Wang Y . Comparison of resistance to powdery mildew and downy mildew in Chinese wild grapes. Vitis 2007; 46: 132.
Xu Y, Zhu Z, Xiao Y, Wang Y . Construction of a cDNA library of Vitis pseudoreticulata native to China inoculated with Uncinula necator and the analysis of potential defence-related expressed sequence tags (ESTs). South Afr J Enol Viticulture 2009; 30: 65–71.
Xu Y, Yu H, He M, Yang Y, Wang Y . Isolation and expression analysis of a novel pathogenesis-related protein 10 gene from Chinese wild Vitis pseudoreticulata induced by Uncinula necator. Biologia 2010; 65: 653–659.
Li H, Xu Y, Xiao Y et al. Expression and functional analysis of two genes encoding transcription factors, VpWRKY1 and VpWRKY2, isolated from Chinese wild Vitis pseudoreticulata. Planta 2010; 232: 1325–1337.
Zhu Z, Shi J, Xu W, Li H, He M, Xu Y, et al. Three ERF transcription factors from Chinese wild grapevine Vitis pseudoreticulata participate in different biotic and abiotic stress-responsive pathways. J of Plant Physiol 2013; 170: 923–933</emph>.
Zhu Z, Shi J, Cao J, He M, Wang Y . VpWRKY3, a biotic and abiotic stress-related transcription factor from the Chinese wild Vitis pseudoreticulata. Plant Cell Rep 2012; 31: 2109–2120.
Zhu Z, Shi J, He M, Cao J, Wang Y . Isolation and functional characterization of a transcription factor VpNAC1 from Chinese wild Vitis pseudoreticulata. Biotechnol Lett 2012; 34: 1335–1342.
Yu Y, Xu W, Wang S et al. VpRFP1, a novel C4C4-type RING finger protein gene from Chinese wild Vitis pseudoreticulata, functions as a transcriptional activator in defence response of grapevine. J Exp Botany 2011; 62: 5671–5682.
YiHe Y, WeiRong X, ShuXiu L, Tao L, YueJin W . Fusion expression and purification of the transcription factor VpRFP1 gene resistance to Uncinula necator from Chinese wild Vitis pseudoreticulata and preparation of its polyclonal antibodies. J Agric Biotechnol 2011; 19: 85–92.
Wen Z, Gao M, Jiao C et al. Characterization and expression analysis of a retinoblastoma-related gene from Chinese wild Vitis pseudoreticulata. Plant Mol Biol Rep 2012; 30: 983–991.
Xu W, Yu Y, Ding J, Hua Z, Wang Y . Characterization of a novel stilbene synthase promoter involved in pathogen-and stress-inducible expression from Chinese wild Vitis pseudoreticulata. Planta 2010; 231: 475–487.
Guan X, Zhao H, Xu Y, Wang Y . Transient expression of glyoxal oxidase from the Chinese wild grape Vitis pseudoreticulata can suppress powdery mildew in a susceptible genotype. Protoplasma 2011; 248: 415–423.
Wen Y, Wang X, Xiao S, Wang Y . Ectopic expression of VpALDH2B4, a novel aldehyde dehydrogenase gene from Chinese wild grapevine (Vitis pseudoreticulata), enhances resistance to mildew pathogens and salt stress in Arabidopsis. Planta 2012; 236: 525–539.
Zhang JK, Sang CG, Wang YJ et al. Expression of powdery mildew resistant genesof chinese wild Vitis by microarray analysis. Acta Hort Sin 2008; 7: 019.
Sanseverino W, Roma G, de Simone M et al. PRGdb: a bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res 2010; 38(Suppl 1): D814–D821.
Tena G, Boudsocq M, Sheen J . Protein kinase signaling networks in plant innate immunity. Curr Opin Plant Biol 2011; 14: 519–529.
Pieterse CM, Leon-Reyes A, van der Ent S, van Wees SC . Networking by small-molecule hormones in plant immunity. Nat Chem Biol 2009; 5: 308–316.
Brutus A, Sicilia F, Macone A, Cervone F, de Lorenzo G . A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc Natl Acad Sci USA 2010; 107: 9452–9457.
Kohorn BD, Kohorn SL . The cell wall-associated kinases, WAKs, as pectin receptors. Front Plant Sci 2012; 3: 88.
Schulze B, Mentzel T, Jehle AK et al. Rapid heteromerization and phosphorylation of ligand-activated plant transmembrane receptors and their associated kinase BAK1. J Biol Chem 2010; 285: 9444–9451.
Shan L, He P, Li J et al. Bacterial effectors target the common signaling partner BAK1 to disrupt multiple MAMP receptor-signaling complexes and impede plant immunity. Cell Host Microbe 2008; 4: 17–27.
Eulgem T, Somssich IE . Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 2007; 10: 366–371.
Birkenbihl RP, Diezel C, Somssich IE . Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol 2012; 159: 266–285.
Bieri S, Mauch S, Shen QH et al. RAR1 positively controls steady state levels of barley MLA resistance proteins and enables sufficient MLA6 accumulation for effective resistance. Plant Cell Online 2004; 16: 3480–3495.
Shen QH, Saijo Y, Mauch S et al. Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. science 2007; 315: 1098–1103.
Decreux A, Messiaen J . Wall-associated kinase WAK1 interacts with cell wall pectins in a calcium-induced conformation. Plant and Cell Physiol 2005; 46: 268–278.
Boudsocq M, Willmann MR, McCormack M et al. Differential innate immune signalling via Ca2+ sensor protein kinases. Nature 2010; 464: 418–422.
Loake G, Grant M . Salicylic acid in plant defence—the players and protagonists. Curr Opin Plant Biol 2007; 10: 466–472.
D’Maris Amick Dempsey AC, Vlot MC, Daniel FK . Salicylic acid biosynthesis and metabolism. Arabidopsis Book 2011; 9: e0156.
Truman W, Bennett MH, Kubigsteltig I, Turnbull C, Grant M . Arabidopsis systemic immunity uses conserved defense signaling pathways and is mediated by jasmonates. Proc Natl Acad Sci USA 2007; 104: 1075–1080.
Doster M, Schnathorst W . Effects of leaf maturity and cultivar resistance on development of the powdery mildew fungus on grapevines. Phytopathology 1985; 75: 318–321.
Faurie B, Cluzet S, Mérillon JM . Implication of signaling pathways involving calcium, phosphorylation and active oxygen species in methyl jasmonate-induced defense responses in grapevine cell cultures. J Plant Physiol 2009; 166: 1863–1877.
Belhadj A, Saigne C, Telef N et al. Methyl jasmonate induces defense responses in grapevine and triggers protection against Erysiphe necator. J Agric Food Chem 2006; 54: 9119–9125.
Makkar HP, Siddhuraju P, Becker K . Plant Secondary Metabolites. Totowa, NJ: Humana Press, 2007.
Schnee S, Viret O, Gindro K . Role of stilbenes in the resistance of grapevine to powdery mildew. Physiol Mol Plant Pathol 2008; 72: 128–133.
Ahuja I, Kissen R, Bones AM . Phytoalexins in defense against pathogens. Trends Plant Sci 2012; 17: 73–90.
Wang Y, Wu J, Park ZY et al. Comparative secretome investigation of Magnaporthe oryzae proteins responsive to nitrogen starvation. J Proteome Res 2011; 10: 3136–3148.
Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E . Simultaneous RNA-Seq analysis of a mixed transcriptome of rice and blast fungus interaction. PloS One 2012; 7: e49423.
Jacobs A, Dry I, Robinson S . Induction of different pathogenesis-related cDNAs in grapevine infected with powdery mildew and treated with ethephon. Plant Pathol 1999; 48: 325–336.
Godfrey D, Able AJ, Dry IB . Induction of a grapevine germin-like protein (VvGLP3) gene is closely linked to the site of Erysiphe necator infection: a possible role in defense? Mol Plant–Microbe Interact 2007; 20: 1112–1125.
Jayasankar S, Li Z, Gray DJ . Constitutive expression of Vitis vinifera thaumatin-like protein after in vitro selection and its role in anthracnose resistance. Funct Plant Biol 2003; 30: 1105–1115.
Monteiro S, Barakat M, Piçarra-Pereira MA, Teixeira AR, Ferreira RB . Osmotin and thaumatin from grape: a putative general defense mechanism against pathogenic fungi. Phytopathology 2003; 93: 1505–1512.
Chang X, Heene E, Qiao F, Nick P . The phytoalexin resveratrol regulates the initiation of hypersensitive cell death in Vitis cell. PLoS One 2011; 6: e26405.
Chang X, Nick P . Defence signalling triggered by Flg22 and Harpin is integrated into a different stilbene output in Vitis cells. PloS One 2012; 7: e40446.
Schweizer P, Vallélian-Bindschedler L, Mösinger E . Heat-induced resistance in barley to the powdery mildew fungus Erysiphe graminis f. sp. hordei. Physiol Mol Plant Pathol 1995; 47: 51–66.
Wei Y, Zhang Z, Andersen CH et al. An epidermis/papilla-specific oxalate oxidase-like protein in the defence response of barley attacked by the powdery mildew fungus. Plant Mol Biol 1998; 36: 101–112.
Ellis J, Dodds P, Pryor T . The generation of plant disease resistance gene specificities. Trends Plant Sci 2000; 5: 373–379.
Caplan JL, Mamillapalli P, Burch-Smith TM, Czymmek K, Dinesh-Kumar S . Chloroplastic protein NRIP1 mediates innate immune receptor recognition of a viral effector. Cell 2008; 132: 449–462.
Lin L, Wang X, Wang Y . cDNA clone, fusion expression and purification of the novel gene related to ascorbate peroxidase from Chinese wild Vitis pseudoreticulata in E. coli. Mol Biol Rep 2006; 33: 197–206.
Jinjin Z, Yuejin W, Xiping W, Keqiang Y, Jinxiao Y . An improved method for rapidly extracting total RNA from Vitis. J Fruit Sci 2003; 3: 005.
Du Z, Zhou X, Ling Y, Zhang Z, Su Z . agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 2010; 38(Suppl 2): W64–W70.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995; 57: 289–300.
Petersen TN, Brunak S, von Heijne G, Nielsen H . SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011; 8: 785–786.
Acknowledgements
This research was supported by the ‘948’ Program, Ministry of Agriculture, China (Grant No. 2011-G21), by the Program for Young Talents in Northwest A&F University (NCET-10-0692, QN2011052) and by a grant from the Program for Innovative Research Team of Grape Germplasm Resource and Breeding (2013KCT-25) to Yan Xu.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Weng, K., Li, ZQ., Liu, RQ. et al. Transcriptome of Erysiphe necator-infected Vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Hortic Res 1, 14049 (2014). https://doi.org/10.1038/hortres.2014.49
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2014.49
This article is cited by
-
Hormone Changes in Tolerant and Susceptible Grapevine Leaves Under Powdery Mildew Infection
Journal of Plant Growth Regulation (2023)
-
Investigation of long non-coding RNAs as regulatory players of grapevine response to powdery and downy mildew infection
BMC Plant Biology (2021)
-
Recent advances in biotechnological studies on wild grapevines as valuable resistance sources for smart viticulture
Molecular Biology Reports (2020)
-
Inferring RPW8-NLRs’s evolution patterns in seed plants: case study in Vitis vinifera
Planta (2020)
-
RNA-seq analysis and fluorescence imaging of melon powdery mildew disease reveal an orchestrated reprogramming of host physiology
Scientific Reports (2019)

{kind=link}
{kind=link}