
Abstract
Next generation sequencing has revolutionized plant biology. Not only has our understanding of plant metabolism advanced using model systems and modern chromatography, but application of ‘omics’-based technology has been widely extended to non-model systems as costs have plummeted and efficiency increased. As a result, important fundamental questions relating to important horticultural crops are being answered, and novel approaches with application to industry are in progress. Here we review recent research advances on development and ripening of fruit crops, how next generation sequencing approaches are driving this advance and the emerging future landscape.
Similar content being viewed by others


Impact of next-generation sequencing (ngs)
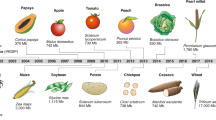
NGS has revolutionized biological research. Since the publication of the Arabidopsis thaliana genome sequence in 2000,1 a deeper understanding of plant biology of both model and crop species has developed by the generation of genome, transcriptome, and epigenome data sets using NGS technology and new bioinformatics algorithms enabling the analysis of resulting data. The rate at which new genomes are being sequenced correlates with the rate at which the cost of the technology has declined (Figure 1). However, software and algorithm development has to some extent lagged behind and shifted limitations from data generation to analysis.2 The plant genomes that have been sequenced to date are shown in Supplementary Table 1. Interestingly, the genomes sequenced in the last five or six years are mainly from species regarded as crops or non-model plants, in contrast with earlier sequencing efforts that concentrated on model plant systems. This change reflects the availability of new technologies and reduced costs as noted above allowing increased applied studies in crops of interest, in addition to more effective use of model systems. Excellent recent reviews focus on advances in genomics related to postharvest physiology and fruit ripening3,4 and impacts of genome sequencing on the research of crops and plants.2,5,6 Here, we focus on NGS technology as a driving force in the advancement of the fields of -omics scale research in fruit crops.

Sequencing cost and sequenced plant genomes this millennia. (a) The cost of sequencing a human genome during the twenty-first century, data uploaded from the National Human Genome Research Initiative (NIH, http://genome.gov/sequencingcosts). (b) Increase in the number of sequenced plant genomes.
Genome sequencing
A direct result of NGS technologies is that sequencing approaches employing BAC-by-BAC-based sequencing methodologies have been replaced by whole-genome shotgun, especially in the last few years with longer read lengths and improved assembly algorithms. It is important to note that the quality of genome sequences can vary depending on the depth of sequence coverage, combinations of sequence methods employed, heterozygosity and complexity of the genome in addition to annotation inputs and methodology, are among the most influential factors. Eighteen fruit genome sequences have been published to date, including grape,7,8 papaya,9 cucumber,10,11 apple,12 strawberry,13 date palm,14 tomato,15 melon,16 banana,17 Chinese plum,18 pear,19,20 watermelon,21 peach,22 kiwifruit23 and pepper.24 However, further progress on genome assembly as well as both physical and functional annotation, which greatly facilitates the use of the information, is needed. The tomato genome assembly is the most complete of all fruit species sequenced so far, but functional annotation of the gene models for this species remains incomplete. The assembly of the apple genome, which was sequenced using a similar strategy to tomato, initially BAC-by-BAC followed by whole-genome shotgun and NGS approaches, has been hampered by heterozygosity and recent gene duplications.12 The assembly of this genome consists of relatively small contigs (averaging ∼7 kb for apple, vs. ∼28 kb for tomato), so building these into a scaffold which is routinely carried out during genome assembly has been difficult, and has led to a high level of redundancy. The estimated number of gene models for apple is relatively high, with 57 386 predicted,12 but recent transcriptome analysis of apple using NGS has revealed an additional 17 524 genes that were not predicted from the original genome assembly.25 Further sequencing and genome assembly is required to better understand the real number of genes encoded in the apple and many plant genomes.
Map-based cloning
The availability of genome sequence enables the possibility for accelerated cloning of genes that characterize a mapped locus. Traditionally, map-based cloning utilizes DNA-based markers that have been mapped to fairly precise areas of plant genomes. Mutants are crossed into other backgrounds over many generations until a small area is fine mapped and can be sequenced using more traditional means, including Sanger sequencing. This approach has been successful for isolation of many ripening mutant loci, especially in tomato,26 as is covered in following sections of this review. In other fruit species, less progress has been made and such studies have focused on candidate genes responsible for ripening-related phenotypes based on their change in expression. The ACC synthase (ACS) alleles in apple have been extensively studied and appear to be involved in shelf-life periods.27–29 However, polygenic mutations are the likely cause of these phenotypes, as the allelic diversity of ACS could not fully explain the full range of shelf-life attributes.30
As well as naturally occurring mutants and variants, collections of insertional, ethylmethane sulfonate (EMS) and fast neutron mutants are available, particularly for tomato.31 The availability of new genomics resources enables identification of the genes responsible for mutant phenotypes at a much faster rate, with a sequenced genome, taking more advantage of fine mapping with fewer inputs due to increased marker diversity. One recent example was the cloning of a GOLDEN-LIKE2 (GLK2) Myb super family transcription factor regulating plastid development and underlying the uniform ripening (u) mutant in tomato.32,33 When a normal U gene is expressed in tomato, dark green areas at the pedicel end of the fruit (shoulders), attributed to higher levels of chlorophyll and numbers of chloroplasts, are prevalent. This mutant has been bred into most commercial cultivars as it results in more even, or ‘uniform’, ripening. Interestingly, U gene expression manifests in a latitudinal gradient in the fruit (higher at the pedicel end compared with the stylar end) opposite to the gradient observed during ripening (from bottom to top) yet consistent with the green shoulder phenotype.33 Transcriptome analysis using RNAseq of these tissues has revealed ripening-related genes that follow a gradient pattern and some novel candidate genes for gradient control were identified, particularly in the more mature stages of ripening.
Functional genomics of fruit ripening
Transcriptional control of ripening
The availability of mutants has been critical to elucidating gene function in plants, and tomato has one of the best collections of characterized fruit ripening-related mutants (maintained and accessible through the Tomato Genetics Resource Center at the University of Davis (http://tgrc.ucdavis.edu/)). Recent reviews describing some of these and the transcriptional control of ripening are available.34–36 In brief, several single locus mutations, resulting in ripening inhibition phenotypes have facilitated understanding of transcriptional control upstream of the required ethylene response for fruit ripening in tomato. Most notable are three ripening mutants, ripening-inhibitor (rin), non-ripening (nor) and colourless non-ripening (Cnr), which show severely inhibited ripening, do not produce elevated endogenous ethylene, or ripen in response to exogenous ethylene, and produce fruit that remain firm and green for extended periods.37,38 However, these mutants show ethylene responsiveness at the level of gene expression indicating they normally control aspects of ripening control in addition to those mediated by ethylene.26 The rin gene was the first cloned,39 and is important not only as a research tool but also commercially. RIN/rin hybrid tomato cultivars delay ripening and extend storage and shelf-life. RIN encodes a MADS-box transcription factor protein of the SEPALATTA clade.40 Cnr is a rare epigenetic mutation, resulting from heritable hyper methylation in the promoter of the Cnr locus SQUAMOSA binding protein (SBP/SPL) causing greatly reduced transcription.38 The expression of CNR is reduced in the rin mutant indicating that it may act downstream of RIN-MADS38 and CNR is also required for RIN binding to target promoters.41
Global effects on ripening pathways, including ethylene independent metabolism, suggest that both RIN and CNR proteins are central to ripening regulation, and conserved through evolution in both climacteric and non-climacteric fruit species. Strawberry is probably the most studied non-climacteric fruit, and a ripening-related MADS-box sequence (FaMADS9)39 was silenced resulting in inhibition of fruit development and ripening.42 This finding provides evidence that transcriptional regulators such as RIN are conserved in both climacteric and non-climacteric species. Furthermore, identification of ripening-related MADS-box genes in maturing banana peel and pulp tissue indicates that RIN function may also be conserved between fleshy fruited monocot and dicot species.43
Ripening control by ethylene
Ethylene is gaseous plant hormone playing roles in many response and developmental processes within plants. The role of ethylene in regulating fruit ripening in climacteric species is well documented.34,36,44 The biosynthetic pathway, perception and signal transduction of ethylene has been described, and the associated genes cloned and characterized in many plant species. Most studies have relied on homology to Arabidopsis sequences in identifying orthologs. This has been very successful, particularly in tomato. The biosynthesis of ethylene starts with the first dedicated and rate-limiting step of the pathway, the conversion of S-adenosyl-l-methionine to ACC by activity of ACS.45 Subsequently ACC is converted to ethylene by activity of ACC oxidase (ACO). The transcriptional regulation of ACS is a major control point of ethylene synthesis,46–48 as it is encoded by a differentially expressed gene family of at least eight in tomato, two of which are upregulated during ripening (LeACS2 and LeACS4).48 At least four family members have been isolated from apple,27,49 of which MdACS1 and MdACS3 have received most attention as they are expressed in fruit.29,50,51 Suppression of ACS using an antisense construct for LeACS2 was successful in tomato.52 ACO is also encoded by a differentially expressed gene family in both tomato and apple: at least four genes exist in tomato, two of which are expressed to high levels during ripening (LeACO1 and LeACO3);53 and at least four genes exist in apple, two of which are expressed in fruit (MdACO1 and MdACO2).50,54,55 Despite ACO genes being transcriptionally upregulated by ethylene and ripening, ACO activity is generally not rate limiting in ripening fruit. Although this is the case, suppression of ACO results in reduced ethylene and delayed ripening in a number of fruit species including tomato, melon, apple, papaya and kiwifruit.56–60 Availability of the tomato genome sequence has led to in silico discovery of a further three putative ACS genes and another three putative ACO genes.15 Functional attributes of these genes remain to be elucidated.
Ethylene signaling starts with its perception by receptors; a multigene family, of seven in tomato.61,62 These receptors are differentially expressed,63,64 three are induced during ripening (LeETR3/Nr, LeETR4 and LeETR6), and are negative regulators of ethylene. In the absence of ethylene, responses are repressed, but in the presence of the hormone, binding to receptors occurs and suppression of the response via a kinase cascade is reversed. Suppression of both LeETR3/Nr and LeETR4 using a fruit-specific RNAi construct resulted in early ripening in transgenic tomato.65 It has also been shown that the receptors are targets of protein degradation via the 26S proteasome pathway64 and that LeETR4 protein is phosphorylated during ripening and exogenous ethylene application.66 Although the exact mechanisms of this protein turnover is not fully understood, it is tempting to speculate that phosphorylation of the receptor labels them for ubiquitination and degradation. Further, it has been observed that both ETR3/Nr and LeETR1 proteins interact with a tetratricopeptide repeat protein (SlTPR1) in a yeast two-hybrid assay.67 These classes of proteins are involved with ubiquitination of targets during 26S proteasome mediated degradation. However, when overexpressed, increased levels of SlTPR1 protein did not cause any ripening-related changes, indicating the requirement of additional regulators in ethylene receptor turnover.64
The identification of the gene underlying the GREEN-RIPE (GR), a fruit-specific and ethylene-insensitive mutation of tomato was the first example of a novel ethylene signaling component discovered in a species other than Arabidopsis.68 Its homolog was also described in Arabidopsis.69 The function of GR is unknown, although its sequence suggests that it is membrane localized and has potential copper binding activities. It has been hypothesized that since the ethylene receptors are bound to the membranes of the endoplasmic reticulum,70,71 and that copper ions are required for their activity, GR may influence copper binding to the receptors.
Following perception of ethylene, downstream signaling takes place via a CONSTITUTIVE TRIPLE RESPONSE1 (CTR) map kinase kinase kinase family of three genes in tomato.72 It has been shown that ETR3/Nr in tomato interacts directly with multiple tomato CTR proteins,73 supporting the hypothesis that ethylene receptors directly transmit a signal to the downstream CTRs. Later steps of ethylene signaling are mediated by the positive regulators ETHYLENE INSENSITIVE 2 (EIN2) and ETHYLENE INSENSITIVE 3 (EIN3). CTR negatively regulates EIN2 and there is just one EIN2 gene in both Arabidopsis and tomato through which all ethylene processes are transmitted. In tomato, EIN2 expression increases at the onset of ripening.74 EIN2 then activates EIN3 encoded by a family of four EIN3-like genes in tomato. Antisense suppression of tomato EIL1, EIL2 and EIL3 reduces ethylene sensitivity. All of these genes are expressed throughout the plant suggesting there is some functional redundancy in tomato.75 A family of two EIN3 BINDING FACTOR (SlEBF1 and SlEBF2) FBOX proteins target the EIN3 protein for degradation via the 26S proteosome and are functionally redundant in tomato. Repression of SlEBF1/SlEBF2 resulted in constitutive ethylene responses and early ripening, indicating that ethylene signaling was activated by the stabilization of the EIL proteins.76 The EILs transcriptionally activate a large family of ETHYLENE RESONSE FACTORS (ERFs), due to the large number of family members some degree of functional redundancy is likely.34
Transcriptomic studies reveal novel regulators of fruit development
There are numerous databases for fruit species where EST sequences have been deposited, and an inventory of these resources for horticultural crops is available.77 These EST sequences have been used for electronic gene expression data sets and for establishing microarrays for analyzing the expression of large numbers of genes in any given sample. However, the target sequence and hybridization limitations of microarrays, combined with the availability of NGS-based RNAseq technologies, has meant that this method has been largely replaced in most systems. Establishing a large-scale picture of gene expression during ripening, or in response to treatment or environmental stress, has become feasible. However, there are still relatively few reports in the ripening literature describing RNAseq, a situation that is likely to change rapidly, especially as no prior sequencing is required for a de novo build of a reference transcriptome, in the absence of an available genome sequence.25,78 In addition, open access tools that provides users a platform for interactive large scale genome analysis further enable genome scale analyses.79 Examples of how transcriptomics has driven the discovery of novel ripening regulators has been especially effective in tomato with numerous examples including LeHB1, TAGL1, FUL1/FUL2, SlAP2 and SlERF6.80–84
Forward genetics has been useful in characterizing non-lethal mutants, but reverse genetics, has enabled the identification of many novel regulators of fruit ripening as noted above. For example, an HD-zip homeobox protein (LeHB1) was identified that binds to the promoter of the ripening-related ACC oxidase, LeACO1.80 Antisense suppression of LeACO1 in tomato results in reduced ethylene production and inhibited ripening.56,85 Previous studies have failed to demonstrate strong RIN binding to the LeACO1 promoter; however, RIN was observed to interact with LeHB1 promoter sequences suggesting a RIN-LeHB1-LeACO1 regulatory pathway in tomato during ripening.41 Putative LeHB1 binding sites are also present on a number of other ripening-related genes including LeACO2, LePG1, MADS-RIN and NOR; therefore, it is possible that LeHB1 might also regulate these ripening-related genes directly.80
Like LeHB1, the Tomato AGAMOUS-like 1 (TAGL1) gene, a ripening-related MADS-box transcription factor of the AGAMOUS clade, was identified through exploration of the tomato genome and its fruit ripening-associated expression profile. TAGL1 is expressed at high levels during early carpel development in addition to ripening associated increase. Suppression of TAGL1 using RNAi led to both ripening inhibition and reduction of carpel thickness due to reduced cell layers,81,86,87 indicating roles in both early and late fruit development associated with both early and late peaks of expression. TAGL1 shares the AGAMOUS clade in tomato with TAG1, the tomato ortholog to the Arabidopsis AGAMOUS gene. TAG1 is expressed in ripening fruit and induced in the absence of TAGL1 expression, indicating possible functional redundancy. TAG1 was shown to have a similar function in carpel development to its Arabidopsis ortholog,88 however, its suppression caused the homeotic conversion of carpels to petals and therefore analysis of fruit development was not possible. However, overexpression of TAG1 caused accumulation of lycopene, and fleshy expansion of sepals88 similar to the ectopic expression of TAGL1 homologs of both tomato and peach.89 The use of fruit specific promoters in the characterization of TAG1 in tomato would be useful to repress its expression without pleiotropic effects on the other aspects of plant development.
In addition to RIN, TAG, and TAGL1, FRUITFUL1 (FUL1; formerly named TDR4) and FUL2 (formerly named MBP7) are MADS-box transcription factors that play a role in regulating ripening.90,91 Silencing of FUL1 results in impaired ripening in tomato, and a ripening-related FUL homolog exists in bilberry.92 Further, it was revealed that FUL1 and FUL2 interact with RIN.82 A transcriptome and ChIP-chip analysis in FUL1/FUL2 suppressed lines identified FUL1/FUL2 target genes that exhibited FUL1/FUL2-dependent expression during ripening. Further, in vitro protein binding assays revealed that RIN, TAGL1, FUL1 and FUL2 form DNA binding complexes suggesting a tetramer complex of these MADS-box proteins are responsible for transcriptional activation of ripening-related genes.82 A bZIP transcription factor from tomato, AREB1, is regulated by abscisic acid and was shown to be expressed during stress93 and in seed and fruit tissues.94 Overexpression of AREB1 resulted in altered primary metabolism indicative of mature fruit, in immature fruit, implicating ripening regulation by abscisic acid.94 A negative regulator of the ripening process was also identified based on its expression profile from a transcriptomic study, SlAP2a, a tomato APETELA2 (AP2) gene family member.83,95 SlAP2a is induced during ripening, however, silencing using RNAi caused higher levels of ethylene and carotenoids culminating in advanced ripening status. It was proposed that SlAP2a functions as a modulator of ripening, thereby providing some balance to the positive action of other ripening regulators such as RIN. 83
Epigenetic control of fruit development and ripening
Epigenetics is a relatively new area of research and can be simply defined as the study of changes in genomes and their activities, not caused by changes in the underlying DNA sequence. Unlike the effects of the DNA sequence (the genotype), changes in gene expression or cellular phenotype mediated by epigenetics are rooted in genome structure, packaging and non-sequence-based modifications. Examples of mechanisms that produce such changes are DNA methylation, acetylation and histone modifications, each of which can influence singularly and together, how genes are expressed without altering the underlying DNA sequence. Such changes may last through cell divisions for the duration of the cell’s life, and may also last for multiple generations even though they do not involve changes in the underlying DNA sequence of the organism, instead, non-genetic factors cause the organism’s genes to behave (or ‘express themselves’) differently. Recently, epigenome dynamics have been shown to occur during fruit development and impact ripening.
There is growing evidence that fruit ripening is influenced by the epigenome. The first evidence was reported by Manning et al.,38 who showed the Cnr mutation in tomato is an epi-allele causing inhibition of ripening through heritable hypermethylation of cytosine residues within the CNR-SPL promoter. Epi-alleles are less stable and as such reversion to wild-type sometimes occurs which manifests as ripened sectors in otherwise non-ripened fruit. A number of whole genome/epigenome scale investigations have subsequently taken place in tomato indicating possible influences on development15 and sexual hybridization.96–98 More recently, epigenome dynamics has been associated with ethylene response and ripening.99 Ripening in climacteric fruit occurs by a developmental change in ethylene response described as System 1 to System 2 ethylene.100 The transition to System 2 ethylene promotes ripening. Mature Green fruit, full size fruit harboring mature seeds, but still a week or so prior to ripening, are able to respond to exogenous ethylene and ripen. However, early during fruit development, System 1 ethylene production is auto inhibitory; fruit of these stages are unable to ripen in response to exogenous ethylene. Treatment of immature fruit with an inhibitor of methyltransferases resulted in ripening of immature fruits well in advance of seed maturation and normal ripening.99 These results suggested that DNA methylation influences the System 1 to System 2 ethylene transition. Whole-genome methylome analysis of tomato fruit at four stages of development from immature to ripe indicated that distinct regions of demethylation were often localized upstream of ripening-related genes.99 In addition, it was observed that these same regions colocalized or were adjacent to the binding sites of the ripening associated transcription factor RIN-MADS protein, as determined by CHiP-seq analysis. In combination, demethylation of ripening gene promoters, RIN–MADS binding and the early ripening of fruit inhibited for methylation, suggests an important role of the epigenome and its dynamics in contributing to ripening control and in particular the control of the transition from System 1 to System 2 ethylene response.
Investigation of the proteome during ripening
Commonly used techniques to identify proteins and map their interactions in a cellular context in plant-based proteomic research includes two-dimensional gel electrophoresis, mass spectrometry (MS), matrix-assisted laser desorption ionization–time of flight (MALDI TOF), yeast two-hybrids screens, ELISA and western blots, complemented in varying degrees with computational prediction programs. Some of these methods have been used for years, but are now being adapted to high throughput screening techniques enabled for improved predictive power by annotated genome sequences and robust computational tools. It is the combined high-throughput screening and increased predictive ability that defines modern proteomics. The accurate quantitation of proteins and peptides in complex biological systems is one of the most challenging areas of proteomics.101 MS-based methodologies have provided significant advances in accurate and sensitive quantitation and the ability to multiplex vastly complex samples through the application of robust bioinformatic tools.101 MS-based methodologies are relatively simple and address the issues of reproducibility102 and under representation of low abundance,103 low mass and basic proteins.104 Accordingly, MS-based methods have come into prominence compared with antibody-based methods due to their higher specificity, good reproducibility and precision, and ability to rapidly analyze hundreds of peptide transitions in a single assay.105
Until recently, most proteomic studies of fruit had been carried out using two-dimensional electrophoresis to separate and identify proteins of interest. MS-based techniques are then used to analyze the sequence of the peptides associated with a ‘picked’ spot. The results of these studies highlight the need for improvements typically low numbers of proteins of interest have been identified and often with a high degree of contamination. These limited proteomes have provided initial proteome insights into fruit ripening and softening, and generally validate prior transcriptomic studies. Comparison of two ripening stages of peach revealed altered abundance of 53 proteins, among these, proteins involved in both primary and secondary metabolism, ethylene biosynthesis, and stress response.106 In a fruit softening study of five peach and nectarine cultivars,107 it was revealed that 164 proteins were differentially changed. Only 14 of these changed during softening in all of the cultivars tested. These proteins were mostly involved in carbohydrate and cell wall metabolism and fruit senescence. In ripening apricot fruit, 106 proteins involved in the biochemical processes influencing metabolic and structural changes during ripening were identified.108 In papaya fruit, 27 ripening-related proteins involved in cell wall metabolism, stress response, ethylene and carotenoid biosynthesis were identified.109 An analysis of the strawberry proteome defined 68 identifiable proteins that were differentially accumulated during ripening, among these, proteins involved with anthocyanin production, heat shock, storage and ripening.110 The proteome of grape revealed changes in photosynthesis, carbohydrate metabolism and stress response at the onset of ripening.111 Further, it was observed that the most significant changes occurred during the first 2 weeks following the onset of ripening, the most notable changes corresponding to pathogen defense, oxidative stress, carbon and nitrogen metabolism.112 Ripening-induced changes in mango revealed 47 differentially expressed proteins, among those involved with carbon fixation, hormone biosynthesis, stress response and pathogen defense.113 In apple, 97 proteins were identified in response to both ripening and ethylene treatment of the following classes: ethylene production, antioxidation and redox, carbohydrate metabolism, oxidative stress, energy and defense response.114 A similar study in apple revealed 53 differentially accumulating proteins involved in stress response and defense, energy and metabolism, fruit ripening and senescence, signal transduction, cell structure and protein synthesis during ripening and storage.115 Proteomic analysis of tomato during ripening in two ecotypes revealed 83 proteins that were differentially expressed involved with redox status, defense, stress, carbon metabolism, energy production and cellular signaling.116 In tomato pericarp, 424 proteins were identified as unique from 12 different genotypes117 and changed in abundance either in response to developmental age (333 proteins), according to genotype (321 proteins) and according to stage and genotype interactions (215 proteins). To isolate the proteome involved in the cuticle of tomato, fruit were dipped in an organic solvent and surface proteins extracted and identified.118 Approximately 200 proteins were identified, a subset of which are potentially involved in the transport, deposition, or modification of cuticle constituents. For a recent review on proteomic investigations on fruit development and ripening see Ref. 119.
The use of iTRAQ (isobaric tag for relative and absolute quantization of tryptic peptides) technology provides a deeper view of the proteome within a sample, and this prompted the development of a bioinformatics pipeline for processing EST data to enable grape peptide prediction.120 Using this peptide database and iTRAQ MS/MS, 76 of 674 identified proteins were shown to be differentially expressed during development and véraison (ripening) of the grape berry, including proteins involved in abiotic and biotic stimuli and sucrose and hexose metabolism.121 A similar investigation was recently carried out in fruit of the Gr non-ripening tomato mutant. Again an iTRAQ MS/MS approach was used and revealed 43 proteins involved in pathways including cell wall metabolism, photosynthesis, oxidative phosphorylation, carbohydrate and fatty acid metabolism, protein synthesis and processing.122 What these, and the above studies suggest, is that fruit development and ripening is a very metabolically active process, involving changes in numerous, pathways related to color, texture, cell wall metabolism, stress response and hormones synthesis.
Metabolomics in ripening
Comprehensive metabolomic studies aim to provide an efficient and accurate determination of the chemical constituents present in a tissue, and are being applied in a large number of biological disciplines.123 Metabolomics technology includes use of nuclear magnetic resonance, and gas- or liquid-chromatography coupled to MS. Fruit is a very rich source of primary and especially secondary metabolites and so was one of the first plant organs to be subjected to investigation. Metabolic studies on ripening of fruit has been carried out in many species including peach, melon, tomato, apple, pear, avocado and pepper.124–130 Overall, these studies have provided new insights into metabolic pathways and sometimes unique chemistry leading to maturation associated changes in appearance, flavor and quality of ripe fruit.
Examples of findings include identification of metabolic networks that underpin each developmental stage during peach fruit development and ripening.124 Early stages were characterized by decreases in protein abundance and elevated levels of polyphenols and amino acids, both substrates for lignin biosynthesis during stone hardening. Sucrose levels increased during development related to translocation from the leaf, and posttranscriptional mechanisms were important for metabolic regulation during early stages of development. Amino-acid levels decreased, coupled with the elevation of transcripts involved with amino acid and organic acid catabolism during ripening stage, consistent with the mobilization of amino acids to support respiration. Sucrose cycling also occurred during storage.
One of the more intriguing recent studies was a comparative investigation of three climacteric (peach and two tomato cultivars) and two non-climacteric species (strawberry and pepper).131 Using a principal component-based analysis, called STASIS, in combination with pathway over enrichment analysis, conserved dynamics of metabolic processes were observed using publically available metabolic profiles for the investigated species. Further, it was demonstrated that this novel computational approach could be used to identify similarities in metabolic processes during fruit development and ripening, thus providing insight into what pathways are essential during these processes across species.
Systems biology
Systems biology is an increasingly important interdisciplinary field, focusing on complex interactions within biological systems, using a holistic rather than a reductionistic approach. Such analyses have been made feasible through the availability of large-scale comprehensive data sets and ever increasing computational power. Typically metabolic, proteomic, transcriptomic, genetic and/or cell signaling networks are generated, making use of mathematical and computational models to find common themes such as correlation among large data sets. One of the major aims of systems biology is to use these models and networks to predict outcomes in the network when perturbed by an environmental cue, treatment, developmental transition or change in genotype. To date, much of this work in fruit has been done in tomato due to the genetic and genomic resources available for this system. As an example, targeted metabolic analysis, coupled with microarray transcriptome profiling, in the ethylene receptor mutant of tomato Nr, revealed multiple points of ethylene control during fruit development.132 It was observed that 869 genes were differentially expressed during normal fruit development, and that the Nr mutation (i.e., ethylene) influenced 37% of these genes. Nr also influenced fruit morphology, seed number, ascorbate accumulation, carotenoid biosynthesis and ethylene evolution, consistent with the ‘well known’ role of ethylene in ripening. Further, 72 of the differentially expressed genes are homologous to known transcription factors and genes involved in signal transduction, indicating complex regulatory control. In a subsequent study, the ripening mutants rin and nor were also compared (in addition to Nr) to wild-type tissues, at the transcriptomic, proteomic, and metabolomic levels during development and ripening.133 Shifts in primary metabolism were revealed by untargeted metabolic profiling, leading to a decrease in metabolic activity during ripening. Combining metabolomics, transcriptomic and proteomic data highlighted several aspects of metabolic control during ripening. Although expression level of transcripts and their corresponding proteins may not always correlate during earlier stages of ripening, much higher correlation was observed during the later stages of ripening, indicating complex posttranscriptional control at the onset and early stages of ripening. Strong correlations were also observed for ripening associated transcripts and specific groups of metabolites including sugars, cell wall-related metabolites and organic acids indicating their importance during the ripening process in tomato. Together these results confirm multiple ethylene-associated events during ripening in tomato. In a comparative systems biology-based study of a climacteric tomato and non-climacteric pepper fruits, both similarities and differences were observed in ripening regulation in these species.130 While both species have similar ethylene-mediated signaling components, their regulation is different indicating possible ethylene insensitivity, or other non-ethylene-mediated regulators in pepper. Ethylene biosynthetic genes were not induced during ripening in pepper, yet genes downstream of ethylene signaling such as carotenoid biosynthesis, cell wall metabolism and the never-ripe receptor were induced in both pepper and tomato.
The use of systems biology in correlation or co-expression-based approaches, have been utilized successfully by the metabolic engineering field to fill gaps in biosynthetic pathways, or to discover novel regulators of biosynthetic pathways. One such example was reported in tomato, where the genetic diversity of the Solanum pennellii introgression lines was exploited to find regulators of carotenoid biosynthesis.84 Transcriptome analysis using the TOM2 microarray and targeted metabolic profiling revealed 953 carotenoid-correlated genes, a subnetwork revealed 38 candidate transcription factors. One of these, SlERF6 was functionally characterized by RNAi and it was revealed that suppression of this gene caused enhanced ethylene and carotenoid synthesis suggesting an important role for SlERF6 in ripening connecting the ethylene and carotenoid pathways.
Translational biology of ripening
As the number of sequenced genomes of fruit species grows, so do opportunities to exploit these genomics resources for translational experiments. The transcriptional regulation of ripening is well characterized in tomato as outlined in previous sections of this review providing opportunities for comparative genomics approaches to identify candidate genes from other fruit species taking advantage of leveraging functional studies performed in tomato. As an example, MADS-box transcription factors play a major role in the transition to and controlling ripening in tomato and other species. In a phylogenetic analysis of the kiwifruit MADS-box transcription factors, mined from the published genome,23 a total of 14 kiwifruit sequences were discovered, and were aligned with other known fruit and Arabidopsis-predicted protein sequences as described previously.43,134 Their expression was confirmed by mining the RNAseq data that were released with the kiwifruit genome.23 Figure 2 shows the phylogenetic relationship and the expression profiles of these 14 putative MADS-box transcription factors from kiwifruit. All were expressed in fruit tissues, however, half of these genes were expressed in low levels in maturing fruit, indicating a role more aligned with floral regulation and early fruit development rather than ripening. The other half of these genes increased during development, either at or following 120 days after pollination indicating a possible role in ripening control. Further, all of the main clades of the tree were represented with at least one kiwifruit gene that was expressed in mature fruit. This approach of mining regulators of ripening provides opportunities for biotechnological and marker-assisted breeding control of ripening.

Phylogeny and expression of putative fruit MADS-box transcription factors. (a) Phylogenetic tree showing the relationship of kiwifruit MADS-box proteins and their homologs from characterized sequences from Arabidopsis, tomato and other fruit species. Bootstrap values are shown on branches, for further details, see Supplementary Data S1. (b) Expression profiles of 14 kiwifruit MADS-box transcription factors.
Future prospects
The pace at which sequencing technologies, computer infrastructure and computational biology has developed has been staggering in the last 20 years. We now have the blueprints of how plants grow and undergo complex developmental processes through the rapidly expanding collection of plant genome sequences. However, interpreting these blueprints remains a challenge. These new ‘omics’-based research tools have already provided novel insights into the underlying biology of fruit development. Opportunities exist to expand this knowledge to postharvest systems, especially to responses of products to treatments such as temperature and modified atmospheres, as technology develops. Earlier this year, Illumina announced the release of their new generation of sequencing platforms, NextSeq 500 and HiSeq X Ten, both promising to lower sequencing costs and the time to generate sequence data. According to Illumina, HiSeq X Ten will yield whole human genome sequences for $1000 each and will have the capability of generating up to 20 000 genomes per year. With these technological advances in mind, the Plant Science Decadal Vision has highlighted ‘Increase the ability to predict plant traits from plant genomes in diverse environments’ and ‘Enhance the ability to find answers in a torrent of data’ as two of its five goals to shape plant biology research in the decade 2015–2025.135 These technical challenges faced will be overcome by the continuing advances in information technology. The prospects for greater understanding of fruit development and the ability to model and predict the effects of the environment during postharvest storage of these fruits look bright.
References
Arabidopsis T, Initiative G . Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000; 408: 796–815.
Hamilton JP, Buell CR . Advances in plant genome sequencing. Plant J 2012; 70: 177–190.
Pech JC, Latché A . Contribution of genomics to postharvest biology. Stewart Postharvest Rev 2013; 9: 1–6.
Pech JC, Purgatto E, Cl G, Cv R, Latché A . Current challenges in postharvest biology of fruit ripening. Curr Agric Sci Technol 2013; 19: 1–18.
Feuillet C, Leach JE, Rogers J, Schnable PS, Eversole K . Crop genome sequencing: lessons and rationales. Trends Plant Sci 2011; 16: 77–88.
Michael TP, Jackson S . The first 50 plant genomes. Plant Genome 2013; 6: 1–7.
Jaillon O, Aury JM, Noel B et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007; 449: 463–467.
Velasco R, Zharkikh A, Troggio M et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2007; 2: e1326.
Ming R, Hou S, Feng Y et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008; 452: 991–996.
Huang S, Li R, Zhang Z et al. The genome of the cucumber, Cucumis sativus L. Nat Genet 2009; 41: 1275–1281.
Wóycicki R, Witkowicz J, Gawroński P et al. The genome sequence of the North-European cucumber (Cucumis sativus L.) unravels evolutionary adaptation mechanisms in plants. PLoS ONE 2011; 6: e22728.
Velasco R, Zharkikh A, Affourtit J et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 2010; 42: 833–839.
Shulaev V, Sargent DJ, Crowhurst RN et al. The genome of woodland strawberry (Fragaria vesca). Nat Genet 2011; 43: 109–116.
Al-Dous EK, George B, Al-Mahmoud ME et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol 2011; 29: 521–527.
Tomato Genome Consortium . The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012; 485: 635–641.
Garcia-Mas J, Benjak A, Sanseverino W et al. The genome of melon (Cucumis melo L.). Proc Natl Acad Sci USA 2012; 109: 11872–11877.
D’Hont A, Denoeud F, Aury JM et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012; 488: 213–217.
Zhang Q, Chen W, Sun L et al. The genome of Prunus mume. Nat Commun 2012; 3: 1318.
Wu J, Wang Z, Shi Z et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res 2013; 23: 396–408.
Chagné D, Crowhurst RN, Pindo M et al. The draft genome sequence of European Pear (Pyrus communis L. ‘Bartlett’). PLoS ONE 2014; 9: e92644.
Guo S, Zhang J, Sun H et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 2013; 45: 51–58.
Verde I, Abbott AG, Scalabrin S et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 2013; 45: 487–494.
Huang S, Ding J, Deng D et al. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun 2013; 4: 2640.
Qin C, Yu C, Shen Y et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 2014; 111: 5135–5140.
Bai Y, Dougherty L, Xu K . Towards an improved apple reference transcriptome using RNA-seq. Mol Genet Genomics 2014; 289: 427–438.
Giovannoni JJ . Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol 2007; 10: 283–289.
Harada T, Sunako T, Sakuraba W et al. Genomic sequence of a ripening related 1-aminocyclopropane-1-carboxylate synthase gene (MdACS-1) in apple (accession no. U89156) (PGR97-066). Plant Physiol 1997; 113: 1465–1466.
Sunako T, Sakuraba W, Senda M et al. An allele of the ripening-specific 1-aminocyclopropane-1-carboxylic acid synthase gene (ACS1) in apple fruit with a long storage life. Plant Physiol 1999; 119: 1297–1304.
Wang A, Yamakake J, Kudo H et al. Null mutation of the MdACS3 gene, coding for a ripening-specific 1-aminocyclopropane-1-carboxylate synthase, leads to long shelf life in apple fruit. Plant Physiol 2009; 151: 391–399.
Bai SL, Wang AD, Igarashi M et al. Distribution of MdACS3 null alleles in apple (Malus×domestica Borkh.) and its relevance to the fruit ripening characters. Breed Sci 2012; 62: 46–52.
Emmanuel E, Levy AA . Tomato mutants as tools for functional genomics. Curr Opin Plant Biol 2002; 5: 112–117.
Powell AL, Nguyen CV, Hill T et al. Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 2012; 336: 1711–1715.
Nguyen CV, Vrebalov JT, Gapper NE et al. Tomato GOLDEN2-LIKE transcription factors reveal molecular gradients that function during fruit development and ripening. Plant Cell 2014; 26: 585–601.
Klee HJ, Giovannoni JJ . Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet 2011; 45: 41–59.
Seymour GB, østergaard L, Chapman NH, Knapp S, Martin C . Fruit development and ripening. Annu Rev Plant Biol 2013; 64: 219–241.
Gapper NE, McQuinn RP, Giovannoni JJ . Molecular and genetic regulation of fruit ripening. Plant Mol Biol 2013; 82: 575–591.
Tigchelaar EC, Mcglasson WB, Franklin MJ . Natural and ethephon-stimulated ripening of F1 hybrids of the ripening inhibitor (rin) and non-ripening (nor) mutants of tomato (Lycopersicon esculentum Mill.). Aust J Plant Physiol 1978; 5: 449–456.
Manning K, Tör M, Poole M et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 2006; 38: 948–952.
Vrebalov J, Ruezinsky D, Padmanabhan V et al. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science 2002; 296: 343–346.
Hileman LC, Sundstrom JF, Litt A, Chen M, Shumba T, Irish VF . Molecular and phylogenetic analyses of the MADS-box gene family in tomato. Mol Biol Evol 2006; 23: 2245–2258.
Martel C, Vrebalov J, Tafelmeyer P, Giovannoni JJ . The tomato MADS-box transcription factor RIPENING INHIBITOR interacts with promoters involved in numerous ripening processes in a COLORLESS NONRIPENING-dependent manner. Plant Physiol 2011; 157: 1568–1579.
Seymour GB, Ryder CD, Cevik V et al. A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria x ananassa Duch.) fruit, a non-climacteric tissue. J Exp Bot 2011; 62: 1179–1188.
Elitzur T, Vrebalov J, Giovannoni JJ, Goldschmidt EE, Friedman H . The regulation of MADS-box gene expression during ripening of banana and their regulatory interaction with ethylene. J Exp Bot 2010; 61: 1523–1535.
Lin Z, Zhong S, Grierson D . Recent advances in ethylene research. J Exp Bot 2009; 60: 3311–3336.
Adams DO, Yang SF . Ethylene biosynthesis: Identification of 1-aminocyclopropane-1-carboxylic acid as an intermediate in the conversion of methionine to ethylene. Proc Natl Acad Sci USA 1979; 76: 170–174.
Rottmann WH, Peter GF, Oeller PW et al. 1-Aminocyclopropane-1-carboxylate synthase in tomato is encoded by a multigene family whose transcription is induced during fruit and floral senescence. J Mol Biol 1991; 222: 937–961.
Nakatsuka A, Murachi S, Okunishi H et al. Differential expression and internal feedback regulation of 1-aminocyclopropane-1-carboxylate synthase, 1-aminocyclopropane-1-carboxylate oxidase, and ethylene receptor genes in tomato fruit during development and ripening. Plant Physiol 1998; 118: 1295–1305.
Barry CS, Llop-Tous MI, Grierson D . The regulation of 1-aminocyclopropane-1-carboxylic acid synthase gene expression during the transition from system-1 to system-2 ethylene synthesis in tomato. Plant Physiol 2000; 123: 979–986.
Rosenfield CL, Kiss E, Hrazdina G . MdACS-2 (accession no. U73815) and MdACS-3 (accession no. U73816): two new l-aminocyclopropane-1-carboxylate synthases in ripening apple fruit. Plant Physiol 1996; 112: 1735–1736.
Wiersma PA, Zhang H, Lu C, Quail A, Toivonen PMA . Survey of the expression of genes for ethylene synthesis and perception during maturation and ripening of “Sunrise” and “Golden Delicious” apple fruit. Postharvest Biol Technol 2007; 44: 204–211.
Zhu Y, Rudell DR, Mattheis JP . Characterization of cultivar differences in alcohol acyltransferase and 1-aminocyclopropane-1-carboxylate synthase gene expression and volatile ester emission during apple fruit maturation and ripening. Postharvest Biol Technol 2008; 49: 330–339.
Oeller PW, Lu MW, Taylor LP, Pike DA, Theologis A . Reversible inhibition of tomato fruit senescence by antisense RNA. Science 1991; 254: 437–439.
Barry CS, Blume B, Bouzayen M, Cooper W, Hamilton AJ, Grierson D . Differential expression of the 1-aminocyclopropane-1-carboxylate oxidase gene family of tomato. Plant J 1996; 9: 525–535.
Dong JG, Olson D, Silverstone A, Yang SF . Sequence of a cDNA coding for a 1-aminocyclopropane-1-carboxylate oxidase homolog from apple fruit. Plant Physiol 1992; 98: 1530–1531.
Ross GS, Knighton ML, Lay-Yee M . An ethylene-related cDNA from ripening apples. Plant Mol Biol 1992; 19: 231–238.
Hamilton AJ, Lycett GW, Grierson D . Antisense gene that inhibits synthesis of the hormone ethylene in transgenic plants. Nature 1990; 346: 284–287.
Ayub R, Guis M, Ben Amor M et al. Expression of ACC oxidase antisense gene inhibits ripening of cantaloupe melon fruits. Nat Biotechnol 1996; 14: 862–6.
Tacken E, Ireland H, Gunaseelan K et al. The role of ethylene and cold temperature in the regulation of the apple POLYGALACTURONASE1 gene and fruit softening. Plant Physiol 2010; 153: 294–305.
Atkinson RG, Gunaseelan K, Wang MY et al. Dissecting the role of climacteric ethylene in kiwifruit (Actinidia chinensis) ripening using a 1-aminocyclopropane-1-carboxylic acid oxidase knockdown line. J Exp Bot 2011; 62: 3821–3835.
Che-Radziah CM, Nurul-Shahnadz AH, Nairatul-Ain AN, Zainal Z . Genetic transformation of antisense ACC oxidase in Carica Papaya L. cv Sekaki via particle bombardment. Malaysian J Appl Biol 2011; 40: 39–45.
Wilkinson JQ, Lanahan MB, Yen HC, Giovannoni JJ, Klee HJ . An ethylene-inducible component of signal transduction encoded by never-ripe. Science 1995; 270: 1807–1809.
Klee HJ . Ethylene signal transduction. Moving beyond Arabidopsis. Plant Physiol 2004; 135: 660–667.
Tieman DM, Taylor MG, Ciardi JA, Klee HJ . The tomato ethylene receptors NR and LeETR4 are negative regulators of ethylene response and exhibit functional compensation within a multigene family. Proc Natl Acad Sci USA 2000; 97: 5663–5668.
Kevany BM, Tieman DM, Taylor MG, Cin VD, Klee HJ . Ethylene receptor degradation controls the timing of ripening in tomato fruit. Plant J 2007; 51: 458–467.
Kevany BM, Taylor MG, Klee HJ . Fruit-specific suppression of the ethylene receptor LeETR4 results in early-ripening tomato fruit. Plant Biotechnol J 2008; 6: 295–300.
Kamiyoshihara Y, Tieman DM, Huber DJ, Klee HJ . Ligand-induced alterations in the phosphorylation state of ethylene receptors in tomato fruit. Plant Physiol 2012; 160: 488–497.
Lin Z, Arciga-Reyes L, Zhong S et al. SlTPR1, a tomato tetratricopeptide repeat protein, interacts with the ethylene receptors NR and LeETR1, modulating ethylene and auxin responses and development. J Exp Bot 2008; 59: 4271–4287.
Barry CS, Giovannoni JJ . Ripening in the tomato Green-ripe mutant is inhibited by ectopic expression of a protein that disrupts ethylene signaling. Proc Natl Acad Sci USA 2006; 103: 7923–7928.
Resnick JS, Wen CK, Shockey JA, Chang C . REVERSION-TO-ETHYLENE SENSITIVITY1, a conserved gene that regulates ethylene receptor function in Arabidopsis. Proc Natl Acad Sci USA 2006; 103: 7917–7922.
Rodríguez FI, Esch JJ, Hall AE, Binder BM, Schaller GE, Bleecker AB . A copper cofactor for the ethylene receptor ETR1 from Arabidopsis. Science 1999; 283: 996–998.
Chen YF, Randlett MD, Findell JL, Schaller GE . Localization of the ethylene receptor ETR1 to the endoplasmic reticulum of Arabidopsis. J Biol Chem 2002; 277: 19861–19866.
Adams-Phillips L, Barry C, Kannan P, Leclercq J, Bouzayen M, Giovannoni J . Evidence that CTR1-mediated ethylene signal transduction in tomato is encoded by a multigene family whose members display distinct regulatory features. Plant Mol Biol 2004; 54: 387–404.
Zhong S, Lin Z, Grierson D . Tomato ethylene receptor–CTR interactions: visualization of NEVER-RIPE interactions with multiple CTRs at the endoplasmic reticulum. J Exp Bot 2008; 59: 965–972.
Wang J, Chen G, Hu Z, Chen X . Cloning and characterization of the EIN2-homology gene LeEIN2 from tomato. DNA Seq 2007; 18: 33–38.
Tieman DM, Ciardi JA, Taylor MG, Klee HJ . Members of the tomato LeEIL (EIN3-like) gene family are functionally redundant and regulate ethylene responses throughout plant development. Plant J 2001; 26: 47–58.
Yang Y, Wu Y, Pirrello J et al. Silencing Sl-EBF1 and Sl-EBF2 expression causes constitutive ethylene response phenotype, accelerated plant senescence, and fruit ripening in tomato. J Exp Bot 2010; 61: 697–708.
Sonah H, Deshmukh RK, Singh VP, Gupta DK, Singh NK, Sharma TR . Genomic resources in horticultural crops: status, utility and challenges. Biotechnol Adv 2011; 29: 199–209.
Alkio M, Jonas U, Declercq M, van Nocker S, Knoche M . Transcriptional dynamics of the developing sweet cherry (Prunus avium L.) fruit: sequencing, annotation and expression profiling of exocarp-associated genes. Hortic Res 2014; 1: 11.
Giardine B . Galaxy: a platform for interactive large-scale genome analysis. Genome Res 2005; 15: 1451–1455.
Lin Z, Hong Y, Yin M, Li C, Zhang K, Grierson D . A tomato HD-Zip homeobox protein, LeHB-1, plays an important role in floral organogenesis and ripening. Plant J 2008; 55: 301–310.
Vrebalov J, Pan IL, Arroyo AJ et al. Fleshy fruit expansion and ripening are regulated by the Tomato SHATTERPROOF gene TAGL1. Plant Cell 2009; 21: 3041–3062.
Fujisawa M, Shima Y, Nakagawa H et al. Transcriptional regulation of fruit ripening by tomato FRUITFULL homologs and associated MADS box proteins. Plant Cell 2014; 26: 89–101.
Chung MY, Vrebalov J, Alba R et al. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J 2010; 64: 936–947.
Lee JM, Joung JG, McQuinn R et al. Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J 2012; 70: 191–204.
Picton S, Barton SL, Bouzayen M, Hamilton AJ, Grierson D . Altered fruit ripening and leaf senescence in tomatoes expressing an antisense ethylene-forming enzyme transgene. Plant J 1993; 3: 469–481.
Itkin M, Seybold H, Breitel D, Rogachev I, Meir S, Aharoni A . TOMATO AGAMOUS-LIKE 1 is a component of the fruit ripening regulatory network. Plant J 2009; 60: 1081–1095.
Pan IL, McQuinn R, Giovannoni JJ, Irish VF . Functional diversification of AGAMOUS lineage genes in regulating tomato flower and fruit development. J Exp Bot 2010; 61: 1795–1806.
Pnueli L, Hareven D, Rounsley SD, Yanofsky MF, Lifschitz E . Isolation of the tomato AGAMOUS gene TAG1 and analysis of its homeotic role in transgenic plants. Plant Cell 1994; 6: 163–173.
Tadiello A, Pavanello A, Zanin D et al. A PLENA-like gene of peach is involved in carpel formation and subsequent transformation into a fleshy fruit. J Exp Bot 2009; 60: 651–661.
Bemer M, Karlova R, Ballester AR et al. The tomato FRUITFULL homologs TDR4/FUL1 and MBP7/FUL2 regulate ethylene-independent aspects of fruit ripening. Plant Cell 2012; 24: 4437–4451.
Shima Y, Fujisawa M, Kitagawa M et al. Tomato FRUITFULL homologs regulate fruit ripening via ethylene biosynthesis. Biosci Biotechnol Biochem. 2014; in press.
Jaakola L, Poole M, Jones MO et al. A SQUAMOSA MADS box gene involved in the regulation of anthocyanin accumulation in bilberry fruits. Plant Physiol 2010; 153: 1619–1629.
Orellana S, Yañez M, Espinoza A et al. The transcription factor SlAREB1 confers drought, salt stress tolerance and regulates biotic and abiotic stress-related genes in tomato. Plant Cell Environ 2010; 33: 2191–2208.
Bastías A, Yañez M, Osorio S et al. The transcription factor AREB1 regulates primary metabolic pathways in tomato fruits. J Exp Bot 2014; 65: 2351–2363.
Karlova R, Rosin FM, Busscher-Lange J et al. Transcriptome and metabolite profiling show that APETALA2a is a major regulator of tomato fruit ripening. Plant Cell 2011; 23: 923–941.
Moxon S, Jing R, Szittya G et al. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res 2008; 18: 1602–1609.
Mohorianu I, Schwach F, Jing R et al. Profiling of short RNAs during fleshy fruit development reveals stage-specific sRNAome expression patterns. Plant J 2011; 67: 232–246.
Shivaprasad PV, Chen HM, Patel K, Bond DM, Santos BACM, Baulcombe DC . A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 2012; 24: 859–874.
Zhong S, Fei Z, Chen YR et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 2013; 31: 154–159.
McMurchie EJ, McGlasson WB, Eaks IL . Treatment of fruit with propylene gives information about the biogenesis of ethylene. Nature 1972; 237: 235–236.
Wasinger VC, Zeng M, Yau Y . Current status and advances in quantitative proteomic mass spectrometry. Int J Proteomics 2013; 2013: 180605.
Lilley KS, Razzaq A, Dupree P . Two-dimensional gel electrophoresis: recent advances in sample preparation, detection and quantitation. Curr Opin Chem Biol 2002; 6: 46–50.
Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R . Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci USA 2000; 97: 9390–9395.
Ly L, Wasinger VC . Protein and peptide fractionation, enrichment and depletion: tools for the complex proteome. Proteomics 2011; 11: 513–534.
Findeisen P, Neumaier M . Mass spectrometry-based clinical proteomics profiling: current status and future directions. Expert Rev Proteomics 2009; 6: 457–459.
Prinsi B, Negri AS, Fedeli C et al. Peach fruit ripening: a proteomic comparative analysis of the mesocarp of two cultivars with different flesh firmness at two ripening stages. Phytochemistry 2011; 72: 1251–1262.
Nilo PR, Campos-Vargas R, Orellana A . Assessment of Prunus persica fruit softening using a proteomics approach. J Proteomics 2012; 75: 1618–1638.
D’Ambrosio C, Arena S, Rocco M et al. Proteomic analysis of apricot fruit during ripening. J Proteomics 2013; 78: 39–57.
Nogueira SB, Labate CA, Gozzo FC, Pilau EJ, Lajolo FM, Oliveira do Nascimento JR . Proteomic analysis of papaya fruit ripening using 2DE-DIGE. J Proteomics 2012; 75: 1428–1439.
Bianco L, Lopez L, Scalone AG et al. Strawberry proteome characterization and its regulation during fruit ripening and in different genotypes. J Proteomics 2009; 72: 586–607.
Deytieux C, Geny L, Lapaillerie D, Claverol S, Bonneu M, Donèche B . Proteome analysis of grape skins during ripening. J Exp Bot 2007; 58: 1851–1862.
Negri AS, Prinsi B, Rossoni M et al. Proteome changes in the skin of the grape cultivar Barbera among different stages of ripening. BMC Genomics 2008; 9: 378.
Andrade Jde M, Toledo TT, Nogueira SB, Cordenunsi BR, Lajolo FM, do Nascimento JRO . 2D-DIGE analysis of mango (Mangifera indica L.) fruit reveals major proteomic changes associated with ripening. J Proteomics 2012; 75: 3331–3341.
Zheng Q, Song J, Campbell-Palmer L et al. A proteomic investigation of apple fruit during ripening and in response to ethylene treatment. J Proteomics 2013; 93: 276–294.
Shi Y, Jiang L, Zhang L, Kang R, Yu ZF . Dynamic changes in proteins during apple (Malus × domestica) fruit ripening and storage. Hort Res 2014; 1: 6.
Rocco M, D’Ambrosio C, Arena S, Faurobert M, Scaloni A, Marra M . Proteomic analysis of tomato fruits from two ecotypes during ripening. Proteomics 2006; 6: 3781–3791.
Xu J, Pascual L, Aurand R et al. An extensive proteome map of tomato (Solanum lycopersicum) fruit pericarp. Proteomics 2013; 13: 3059–3063.
Yeats TH, Howe KJ, Matas AJ, Buda GJ, Thannhauser TW, Rose JK . Mining the surface proteome of tomato (Solanum lycopersicum) fruit for proteins associated with cuticle biogenesis. J Exp Bot 2010; 61: 3759–3771.
Palma JM, Corpas FJ, del Río LA . Proteomics as an approach to the understanding of the molecular physiology of fruit development and ripening. J Proteomics 2011; 74: 1230–1243.
Lücker J, Laszczak M, Smith D, Lund ST . Generation of a predicted protein database from EST data and application to iTRAQ analyses in grape (Vitis vinifera cv. Cabernet Sauvignon) berries at ripening initiation. BMC Genomics 2009; 10: 50.
Kambiranda D, Katam R, Basha SM, Siebert S . iTRAQ-based quantitative proteomics of developing and ripening muscadine grape berry. J Proteome Res 2014; 13: 555–569.
Pan X, Zhu B, Zhu H et al. iTRAQ protein profile analysis of tomato Green-ripe mutant reveals new aspects critical for fruit ripening. J Proteome Res 2014; 13: 1979–1993.
Hanhineva K, Aharoni A . Metabolomics in fruit development. In: Jain SM Brar D (eds.). Molecular Techniques in Crop Improvement. Amsterdam: Springer, 2009: 675–693.
Lombardo VA, Osorio S, Borsani J et al. Metabolic profiling during peach fruit development and ripening reveals the metabolic networks that underpin each developmental stage. Plant Physiol 2011; 157: 1696–1710.
Moing A, Aharoni A, Biais B et al. Extensive metabolic cross-talk in melon fruit revealed by spatial and developmental combinatorial metabolomics. New Phytol 2011; 190: 683–696.
Oms-Oliu G, Hertog MLATM, van de Poel B, Ampofo-Asiama J, Geeraerd AH, Nicolaï BM . Metabolic characterization of tomato fruit during preharvest development, ripening, and postharvest shelf-life. Postharvest Biol Technol 2011; 62: 7–16.
Rudell DR, Mattheis JP, Hertog MLATM . Metabolomic change precedes apple superficial scald symptoms. J Agric Food Chem 2009; 57: 8459–8466.
Pedreschi R, Franck C, Lammertyn J et al. Metabolic profiling of ‘Conference’ pears under low oxygen stress. Postharvest Biol Technol 2009; 51: 123–130.
Pedreschi R, Muñoz P, Robledo P et al. Metabolomics analysis of postharvest ripening heterogeneity of “Hass” avocadoes. Postharvest Biol Technol 2014; 92: 172–179.
Osorio S, Alba R, Nikoloski Z, Kochevenko A, Fernie AR, Giovannoni JJ . Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiol 2012; 159: 1713–1729.
Klie S, Osorio S, Tohge T et al. Conserved changes in the dynamics of metabolic processes during fruit development and ripening across species. Plant Physiol 2014; 164: 55–68.
Alba R, Payton P, Fei Z et al. Transcriptome and selected metabolite analyses reveal multiple points of ethylene control during tomato fruit development. Plant Cell 2005; 17: 2954–2965.
Osorio S, Alba R, Damasceno CMB et al. Systems biology of tomato fruit development: combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (Nr) mutants reveals novel regulatory interactions. Plant Physiol 2011; 157: 405–425.
Ireland HS, Yao JL, Tomes S et al. Apple SEPALLATA1/2-like genes control fruit flesh development and ripening. Plant J 2013; 73: 1044–1056.
Martin C . The plant science decadal vision. Plant Cell 2013; 25: 4773–4774.
Acknowledgements
The authors would like to thank Dr Zhangjun Fei for supplying the kiwifruit RNAseq data. Work in our labs was supported by grants from, NSF Plant Genome Research Program, USDA-ARS and Federal Formula Funds project NE1336.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Supplementary Data S1.
FASTA file of aligned MADS box transcription factors and alignment. (DOCX 21 kb)
Supplementary Table S1.
GenBank accession numbers for MADS-box proteins included in phylogenetic analyses. MDP numbers are for apple gene models available from the Genome Database for Rosaceae (www.rosaceae.org). (DOCX 13 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Gapper, N., Giovannoni, J. & Watkins, C. Understanding development and ripening of fruit crops in an ‘omics’ era. Hortic Res 1, 14034 (2014). https://doi.org/10.1038/hortres.2014.34
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2014.34
This article is cited by
-
A MADS-box gene, EgMADS21, negatively regulates EgDGAT2 expression and decreases polyunsaturated fatty acid accumulation in oil palm (Elaeis guineensis Jacq.)
Plant Cell Reports (2020)
-
Re-analysis of long non-coding RNAs and prediction of circRNAs reveal their novel roles in susceptible tomato following TYLCV infection
BMC Plant Biology (2018)
-
Comparative transcriptome analysis reveals distinct ethylene–independent regulation of ripening in response to low temperature in kiwifruit
BMC Plant Biology (2018)
-
High-resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening
Nature Communications (2018)
-
Metabolic Reprogramming in Leaf Lettuce Grown Under Different Light Quality and Intensity Conditions Using Narrow-Band LEDs
Scientific Reports (2018)

{kind=link}