
Abstract
Spondyloepiphyseal dysplasia congenita (SEDC) is an extremely rare autosomal dominant chondrodysplasia that is usually caused by substitution of glycine with another amino acid in the triple helical region of COL2A1. Herein, we describe a case of SEDC in a Chinese family with a novel de novo mutation in the COL2A1 gene, c.1150G>A (p.Gly384Ser), which may impair protein stability and lead to dysfunction of type II collagen.
Similar content being viewed by others


Introduction
Spondyloepiphyseal dysplasia congenita (SEDC, OMIM 183900) is a rare autosomal dominant inherited chondrodysplasia, which was first described by Spranger and Wiedemann in 1966.1 The most common features of SEDC are skeletal deformities such as short-trunk dwarfism, odontoid hypoplasia, cervical spine subluxation, scoliosis, kyphosis, lumbar lordosis, coxa vara, genu valgum, clubfoot, pes planus and metaphyseal changes. Extraskeletal features, including mid-face hypoplasia, sensorineural hearing loss, ocular complications, cleft palate, micrognathia, thoracic hyperkyphosis and hypoplastic abdomen, have also been reported in some cases.2 In 1980’s, studies found a causative association between the abnormal mobility of type II collagen and SEDC, and heterozygous mutations in COL2A1 have since been identified in patients with SEDC phenotypes.
SEDC is a rare disease with a prevalence of 3.4/1,000,000, which mostly results from random mutations sparsely distributed in the 54 exons of the COL2A1 gene. To date, a total of 539 different mutations have been identified globally and listed in the Human Gene Mutation Database, including 283 missense/nonsense mutations, 99 splicing mutations, 101 small deletions, 32 small insertions, 9 small indels, 11 gross deletions, 2 gross insertions and 2 complex rearrangements (BIOBASE Human Gene Mutation Database professional 2017.1, http://www.hgmd.cf.ac.uk/ac/index.php). So far, at least 56 distinct mutations related to SEDC have been reported in different ethnic groups.3 However, the relationship between genomic mutations and their corresponding phenotypes in SEDC remains unclear. The most common causative mutation of SEDC (74%) is a single base substitution in the glycine residue of the triple-helical region of COL2A1, and a change from arginine to cysteine accounts for another 10% of causative mutations.4 Some glycine to serine substitutions (e.g., p.Gly504Ser) result in milder skeletal dysplasia, although glycine to non-serine residue substitutions cause varying phenotypic abnormalities.4,5 The most frequent mutation, p.Arg989Cys, is associated with severe SEDC phenotypes.6,7
In the current study, we describe a novel de novo mutation (c.1150G>A, p.Gly384Ser) in COL2A1, which causes SEDC. The mutation may impair protein stability and lead to dysfunction of type II collagen, and may therefore be pathogenic. Our study supplements the known spectrum of SEDC mutations, may contribute to a better understanding of the genotype–phenotype correlation, and might be helpful in for the genetic counseling of patients with SEDC.
The patient was a boy with non-consanguineous Chinese parents. His family members, including his parents and his elder sister, were apparently healthy and did not display any symptoms or signs of SEDC. There was no family history of skeletal dysplasia. Short stature was noted after birth. Although his psychomotor development was normal, his short stature became more evident as he grew, and skeletal abnormalities were noted including scoliosis, pectus carinatum and metaphyseal changes. At 6 years and 11 months of age, his height was 106 cm (<3rd percentile for normal Chinese males), and his weight was 21 kg (<25th percentile). His hearing was not impaired. Routine blood and urine tests were normal. The analysis of enzyme activity related to lysosomal storage diseases was also normal.
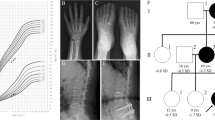
Radiographic examination revealed mild kyphosis and severe lumbar lordosis, flattened and irregular vertebral bodies, wedge-shaped lumbar vertebral bodies, flattening of the acetabular roof and bilateral dysplasia of the femoral heads (Figure 1). Based on these clinical and radiological findings, the patient was believed to have type II collagenopathy, SEDC.

Radiographs of the SEDC patient in our study (a–c), Spinal and pelvic radiographs show flattened and irregular vertebral bodies, lumbar lordosis, wedge-shaped lumbar vertebral bodies, flattening of the acetabular roof and bilateral dysplasia of the femoral heads.
We obtained written informed consent from his parents to perform molecular studies, which were approved by the Institutional Review Board of Peking University First Hospital. We performed whole exome sequencing of a genomic DNA sample from the proband (II:2) of the Chinese Han family with SEDC (Figure 2a). A total of 141,178 genetic variants, including 12,547 non-synonymous changes, were identified in coding sequences or in the canonical dinucleotide of splice site junctions. Variants were functionally annotated and filtered using our cloud-based rare disease NGS analysis platform (https://www.gene.ac/), as previously described.8 Exonic sequence alterations and intronic variants at exon-intron boundaries, with unknown frequency or minor allele frequency<1%, and not present in the homozygous state in those databases, were retained. Among them, 66 variants were found to be associated with skeletal disorders. In total 15 variants were associated with a dominant inheritance mode. Subsequently, a missense mutation in the COL2A1 gene, with a G to A transition at position 1,150, resulting in a substitution of glycine for serine at amino acid position 384 (c.1150G>A, p.Gly384Ser) in the Gly-X-Y triple helical repeating motifs of COL2A1, was identified as the potential SEDC-causing mutation (Figure 2b). This variant has not been described in any other databases, including dbSNP, OMIM, ESP, ClinVar, 1000 Genomes, Human Gene Mutation Database, gnomAD and ExAc.

The pedigree, sequence maps and conservation analysis of COL2A1 with de novo mutation. (a) sequencing chromatograms show a de novo heterozygous mutation, c.1150G>A, in the COL2A1 gene. The red arrow indicates the position of the nucleotide mutation. (b) cross-species protein conservation of COL2A1 around the amino acid alteration, p.Gly384Ser, is displayed. The highly conserved Gly-X-Y triplet sequence is observed in different species. The black box shows the mutation site, G384.
After validation by Sanger sequencing, this variation was observed only in the proband but not in his family members (Figure 2a). Primer sequences used for validating the causative COL2A1 gene variant were as follows: F: 5′- AGAAAAACGGCAGCGTGAAC-3′ and R: 5′- AGAAGCTGCACTTACGGAGG-3′.
Three online software programs were used to predict the functional effects of this amino acid substitution. According to PolyPhen-2, this de novo mutation in our study is predicted to probably be damaging with a score of 0.999 (sensitivity: 0.14, specificity: 0.99) (http://genetics.bwh.harvard.edu/pph2/).9 Using another program, PROVEAN, we predicted p.Gly384Ser to be damaging with a PROVEAN score of −4.567, where scores below −2.5 are deleterious (http://provean.jcvi.org/index.php).10 MutationTaster also predicted that the alteration was disease-causing (http://www.mutationtaster.org/).11 All of these computer-based protein analyses indicate that the de novo mutation of COL2A1 gene was likely the deleterious disease-causing mutation in this patient. We therefore concluded that the de novo mutation, c.1150G>A, was very likely to be the major cause of SEDC in this patient.
SEDC is an autosomal dominant genetic chondrodysplasia resulting from pathogenic mutations in the COL2A1 gene encoding collagen II, which predominantly contributes to the fibrillar matrix of articular cartilage. The COL2A1 gene is >30 kb in length with 54 exons, and encodes a 134.4 kDa protein with 1487 amino acids. Collagen II has three domains: an N-propeptide which may be involved in the regulation of primary fibril diameters, a triple-helical domain which contains 330 Gly-X-Y repeats and is the predominant motif and a C-propeptide which is thought to play a fundamental role in the initiation of triple helix formation. After being secreted into the extracellular matrix, the N- and C-propeptides are cleaved to form the mature type II collagen.12
The group of Andrzej Fertala has demonstrated that some mutations in COL2A1 alter individual collagen molecules and subsequently the structure of collagen fibrils, which has a negative impact on binding partners, resulting in a decrease in the thermostability of collagen. Furthermore, some mutations result in a slow rate of secretion into the extracellular space, and the accumulated structurally changed molecules activate an unfolded protein response and increase the apoptosis of host cells.7,13–15 Experiments in SEDC mice showed skeletal and growth plate abnormalities, along with impaired hearing and retinoschisis.16,17 Further analysis indicated that mutant collagen was unable to provide the normal meshwork required for matrix integrity and overall cartilage stability,18 because this mutant collagen was largely synthesized and retained in the rough endoplasmic reticulum (RER). As a result, apoptosis of chondrocytes was induced by activation of the endoplasmic reticulum stress (ERS)-unfolded protein response (UPR)-apoptosis cascade.19 Histological studies performed on type II collagen extracted from the femoral head cartilage of new type II collagenopathy patients by Su et al.20 suggested that the expression and distribution of collagen II were abnormal, resulting in pathological changes of the embedded chondrocytes and abnormalities in the hierarchical structure of cartilage. Moreover, ultrastructural studies showed aberrant nuclei and RER in mutant chondrocytes, and disarranged collagen fibers in mutant cartilage.20
This report describes a patient with an SEDC phenotype and a novel de novo COL2A1 mutation. Three computer-based protein analyses, PolyPhen2, PROVEAN and MutationTaster, indicated that this mutation was likely to be damaging, and we therefore concluded that the p.Gly384Ser mutation was pathogenic. Until now, including the p.Gly384Ser mutation, at least 57 different mutations in the COL2A1 gene have been described globally to be related to SEDC, with a range of phenotypes. However, the relationships between these mutations and their corresponding clinical manifestations are far from clear, and the expression profiles and characteristics of these mutant proteins still need to be explored.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Spranger J, Wiedemann HR . Dysplasia spondyloepiphysaria congenita. Helv Paediatr Acta 1966; 21: 598.
Deng H, Huang X, Yuan L . Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res 2016; 768: 1–13.
Liu L, Pang Q, Jiang Y, Li M, Wang O, Xia W . Novel COL2A1 mutations causing spondyloepiphyseal dysplasia congenita in three unrelated Chinese families. Eur Spine J 2016; 25: 2967–2974.
Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D et al. Mutation Update for COL2A1 Gene variants associated with type II collagenopathies. Hum Mutat 2016; 37: 7–15.
Kawano O, Nakamura A, Morikawa S, Uetake K, Ishizu K, Tajima T . Spondyloepiphyseal dysplasia congenita caused by double heterozygous mutations in COL2A1. Am J Med Genet A 2015; 167: 1578–1581.
Silveira KC, Bonadia LC, Superti-Furga A, Bertola DR, Jorge AA, Cavalcanti DP . Six additional cases of SEDC due to the same and recurrent R989C mutation in the COL2A1 gene–the clinical and radiological follow-up. Am J Med Genet A 2015; 167A: 894–901.
Steplewski A, Ito H, Rucker E, Brittingham RJ, Alabyeva T, Gandhi M et al. Position of single amino acid substitutions in the collagen triple helix determines their effect on structure of collagen fibrils. J Struct Biol 2004; 148: 326–337.
Chen D, Zhao N, Wang J, Li Z, Wu C, Fu J et al. Whole-exome sequencing analysis of Waardenburg syndrome in a Chineses family. Hum Genome Var 2017; 4: 17027.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP . Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012;. 7: e46688.
Schwarz JM, Cooper DN, Schuelke M, Seelow D . MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361–362.
Gelse K, Poschl E, Aigner T . Collagens—structure, function, and biosynthesis. Adv Drug Deliv Rev 2003; 55: 1531–1546.
Chung HJ, Jensen DA, Gawron K, Steplewski A, Fertala A . R992C (p.R1192C) Substitution in collagen II alters the structure of mutant molecules and induces the unfolded protein response. J Mol Biol 2009; 390: 306–318.
Ito H, Rucker E, Steplewski A, McAdams E, Brittingham RJ, Alabyeva T et al. Guilty by association: some collagen II mutants alter the formation of ECM as a result of atypical interaction with fibronectin. J Mol Biol 2005; 352: 382–395.
Steplewski A, Majsterek I, McAdams E, Rucker E, Brittingham RJ, Ito H et al. Thermostability gradient in the collagen triple helix reveals its multi-domain structure. J Mol Biol 2004; 338: 989–998.
Donahue LR, Chang B, Mohan S, Miyakoshi N, Wergedal JE, Baylink DJ et al. A missense mutation in the mouse Col2a1 gene causes spondyloepiphyseal dysplasia congenita, hearing loss, and retinoschisis. J Bone Miner Res 2003; 18: 1612–1621.
Sahlman J, Pitkanen MT, Prockop DJ, Arita M, Li SW, Helminen HJ et al. A human COL2A1 gene with an Arg519Cys mutation causes osteochondrodysplasia in transgenic mice. Arthritis Rheum 2004; 50: 3153–3160.
Macdonald DW, Squires RS, Avery SA, Adams J, Baker M, Cunningham CR et al. Structural variations in articular cartilage matrix are associated with early-onset osteoarthritis in the spondyloepiphyseal dysplasia congenita (sedc) mouse. Int J Mol Sci 2013; 14: 16515–16531.
Liang G, Lian C, Huang D, Gao W, Liang A, Peng Y et al. Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a col2a1 p.Gly1170Ser mutated mouse model. PLoS ONE 2014; 9: e86894.
Su P, Zhang L, Peng Y, Liang A, Du K, Huang D . A histological and ultrastructural study of femoral head cartilage in a new type II collagenopathy. Int Orthop 2010; 34: 1333–1339.
Data Citations
Yang, Yanling, & Xiao, Han HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1741 (2017)
Acknowledgements
We thank all patients and control individuals for their generous participation in this study. We thank Dr Xiyuan Li for expert NGS data analysis. This work is sponsored by the Fund for Shanxi “1331 Project” Collaborative Innovation Centre, 1331 CIC (206541001).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Xiong, Q., Liu, Y., Xue, Y. et al. A novel de novo mutation in COL2A1 leading to spondyloepiphyseal dysplasia congenita in a Chinese family. Hum Genome Var 5, 17059 (2018). https://doi.org/10.1038/hgv.2017.59
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.59
