
Abstract
Alpha-thalassemia/mental retardation syndrome X-linked (ATRX; OMIM #301040), which is caused by mutations in the ATRX gene, is characterized by alpha-thalassemia, distinct dysmorphic facies, psychomotor development delay and genital abnormalities. Here, we describe a neonatal case of syndromic disorder of sex development, harboring a novel hemizygous mutation, p.Asp2352fs*1 in the carboxyl-terminal domain of ATRX. Our study provides additional evidence that deletion of the carboxyl terminus of ATRX is associated with severe genital anomalies.
Similar content being viewed by others

Alpha-thalassemia/mental retardation syndrome X-linked (ATRX; OMIM #301040) is associated with alpha-thalassemia, distinct dysmorphic facies, psychomotor development delay and genital abnormalities; this syndrome results from mutations in the ATRX gene.1,2 In addition to skeletal anomalies, renal and cardiac anomalies are also present. Genital abnormalities are observed in 80% of male ATRX patients,3 ranging from mild, as in undescended testes, to ambiguous or female phenotypes with streak gonads; thus ATRX is a significant malformation syndrome causing disorder of sex development (DSD) during the neonatal period.4 Here, we describe a neonatal case of syndromic DSD, harboring a novel hemizygous, p.Asp2352fs*1 mutation in the carboxyl-terminal domain of ATRX.
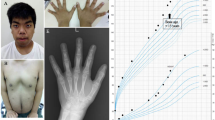
The propositus was a 5-month-old Japanese boy born at 36 weeks of gestation after an uncomplicated pregnancy and delivery. The parents were nonconsanguineous and phenotypically normal. He was the first child of the parents. His birth weight was 2,102 g (−2.1 s.d.), length was 45.0 cm (−1.9 s.d.), and his occipital frontal circumference was 32.0 cm (−0.6 s.d.). The Apgar scores were 5 and 6 at 1 and 5 min, respectively. Dyspnea ensued shortly after birth, and he required positive airway pressure support for a few days. At birth, several dysmorphic features including a dysmorphic face (depressed nasal bridge, hypertelorism, micrognathia and low-set ear), ambiguous genitalia (Figure 1a), polydactyly of the right foot (Figure 1b) and ventriculoseptal defect (detected by echocardiography) were observed. Ultrasonography and magnetic resonance imaging revealed that the bilateral testes were small but detectable (right: 0.7×0.5 cm, left: 0.8×0.6 cm) in the inguinal canals; there were no Müllerian structures. No apparent skeletal deformity was documented on X-ray, other than polydactyly of the right foot. An auditory brain stem response examination revealed hearing loss: 60 dB on the left side and no response on the right side at the age of 2 months. His eyes were examined by experienced ophthalmologists, which revealed no ocular malformation.

Clinical features of the patient. (a) The patient showed ambiguous genitalia at 4 months of age. Micropenis (penile length was 1.9 cm) with penoscrotal hypospadias was observed. No masses were palpable bilaterally. There was no skin hyperpigmentation. (b) Photographs of the feet. The right foot had two additional toes.
Owing to the ambiguous genitalia with micropenis and penoscrotal hypospadias presented by the patient, he was referred to us for hormonal evaluation at the age of 4 months. His weight was 3,960 g (−3.7 s.d.) and length was 54.0 cm (−4.4 s.d.). The basal testosterone concentration was 1.55 ng/ml and, after stimulation, increased to 3.15 ng/ml (Table 1). The secretions of luteinizing hormone and follicle-stimulating hormone in response to gonadotropin-releasing hormone was increased (Table 1) during ‘mini-puberty,’ indicating primary hypogonadism. His serum concentration of 7-dehydrocholesterol was not elevated (0.4 μg/ml, ref: 0.3–2.0). He was not anemic (hemoglobin concentration was 16.5 g/dl). His G-banded karyotype was 46,XY.
We performed whole-exome sequencing to investigate the etiology of syndromic DSD in this patient. After gaining approval from the Institutional Review Board of Keio University School of Medicine and informed consent from the family of the patient, we collected genomic DNA from the patient’s peripheral blood leukocytes. The DNA used in the whole-exome sequencing was collected using a SureSelect Human All ExonV5 Kit (Agilent Technologies, Santa Clara, CA, USA) and was sequenced on a HiSeq 2500 sequencer (Illumina, San Diego, CA, USA) with 100 bp paired-end reads. The methods used herein have been described in detail previously.5 We identified a novel hemizygous frameshift mutation, c.7054delG, p.Asp2352fs*1, in ATRX (Figure 2). We confirmed the mutation detected in the patient by Sanger sequencing. This mutation was absent from the databases, including the dbSNP, 1000 Genomes Project, Exome Variant Server, NHLBI Exome Sequencing Project and Human Genetic Variation Database for the Japanese population. No other mutations associated with DSD (genes associated with DSD are listed in Supplementary Table) were identified. Parental analysis was refused.

Identification of sequence variation in ATRX. Partial sequence of PCR product and schematic diagrams of ATRX protein. The chromatogram shows a hemizygous 1 base pair deletion. The p.Asp2352fs*1 mutation results in the lack of only the carboxyl-terminal domain, whereas other functional domains, such as the amino-terminal ATRX–DNMT3–DNMT3L (ADD) domain and helicase/ATPase domain remain intact. The P-box is an element conserved among other SNF2-like family members involved in transcriptional regulation, and a stretch of glutamine residues (Q-box) represents a potential protein interaction domain.
The ATRX protein belongs to the family of SWI/SNF DNA helicases, which are involved in chromatin remodeling.6 The p.Asp2352fs*1 mutation results in the lack of only the carboxyl-terminal domain, whereas other functional domains, such as the amino-terminal ATRX–DNMT3–DNMT3L (ADD) domain, which is a plant homeodomain-like zinc finger with an additional C2–C2 motif, and the helicase/adenosine triphosphatase (ATPase) domain, which comprises seven highly conserved collinear helicase motifs, remain intact. To date, more than 150 mutations of ATRX have been described. The majority of mutations reported so far in ATRX are missense mutations (about 100 missense mutations have been reported), and clustered in the two ADD and helicase domains.7,8 Mutations in the ADD domain are significantly associated with more severe psychomotor impairment than mutations in the helicase domain.9 The carboxyl terminus of ATRX encodes two additional domains of potential functional importance. The P-box is an element conserved among other SNF2-like family members involved in transcriptional regulation, and a stretch of glutamine residues (Q-box) represents a potential protein interaction domain.3,10 Of interest, it has been reported that in the absence of this carboxyl-terminal domain, severe urogenital abnormalities occur, such as female phenotype with streak gonads or ambiguous genitalia,7 as was observed in our case. It appears likely that this region plays a specific role in urogenital development.
Theoretically, the messenger RNA with c.7054delG seems to result in the nonsense-mediated messenger RNA decay, because this frameshift occurs in the third last exon of ATRX. However, previous studies have shown that some truncating mutations in ATRX with premature stop codon escape from nonsense-mediated messenger RNA decay, and truncated protein was seen in western blotting with protein extracts derived from cell lines carrying these mutations.7 Further, some full-length ATRX protein has been identified in protein extracts derived from cell lines carrying these truncating mutations.7 Therefore, the mutant Asp2352fs*1 ATRX protein could be expressed to some extent, which results in the ambiguous genitalia in our case.
Alpha-thalassemia is one of the defining elements of this syndrome (85% in cases) and is a useful reminder that a simple investigation can be used to establish a diagnosis. However, it is well known that there is considerable variation in the hematologic manifestations associated with ATRX mutations.7 A diagnosis of ATRX in a neonate without anemia essentially depends on facial gestalt, DSD in males and psychomotor development delay combined with other nonspecific findings. Facial dysmorphism, such as telecanthus or widely spaced eyes, short nose, tented vermilion of the upper lip, and thick or everted vermilion of the lower lip, are too subtle to be easily identified, particularly at younger ages. Therefore, the diagnosis of ATRX without anemia based only on clinical manifestation is not necessarily easy in a neonate. However, the early diagnosis of ATRX is important for medical intervention and follow-up, as many children with ATRX have gastrointestinal symptoms,11–13 such as profound gastroesophageal reflux, which may lead to aspiration pneumonia and is a known cause of death in this syndrome, difficulties in feeding manifested often by abdominal pain, and distention and the patient’s refusal for food, along with DSD and intellectual disability. Through whole-exome sequencing, we successfully identified a causative mutation in ATRX in this male patient, who did not exhibit anemia.
In summary, we described a patient with syndromic DSD harboring a novel hemizygous mutation in ATRX. Our study provides additional evidence that deletion of the carboxyl terminus of ATRX is associated with severe genital anomalies. Because whole-exome sequencing enables the screening of most protein-coding genes in an unbiased manner, this technique may help to identify atypical or previously unappreciated phenotypes associated with a known disease-associated gene.
References
References
Gibbons RJ, Brueton L, Buckle VJ, Burn J, Clayton-Smith J, Davison BCC et al. Clinical and hematological aspects of the X-linked a-thalassemia/mental retardation syndrome (ATR-X). Am J Med Genet 1995; 55: 288–299.
Gibbons RJ, Picketts DJ, Villard L, Higgs DR . Mutations in a putative global transcriptional regulator cause X-linked mental retardation with a-thalassemia (ATR-X syndrome). Cell 1995; 80: 837–845.
Gibbons RJ, Higgs DR . Molecular-clinical spectrum of the ATR-X syndrome. Am J Med Genet 2000; 97: 204–212.
Hutson JM, Grover SR, O'Connell M, Pennell SD . Malformation syndromes associated with disorders of sex development. Nat Rev Endocrinol 2014; 10: 476–487.
Takagi M, Shimizu M, Suzuki E, Shinohara H, Narumi S, Hasegawa T et al. Whole exome sequencing identified a novel COL2A1 mutation that causes mild spondylo-epiphyseal dysplasia mimicking autosomal dominant brachyolmia. Am J Med Genet A 2016; 170: 795–798.
Lacoste C, Leheup B, Agouti I, Mowat D, Giuliano F, Badens C . Mutations of codon 2085 in the helicase domain of ATRX are recurrent and cause ATRX syndrome. Clin Genet 2014; 86: 502–503.
Gibbons RJ, Wada T, Fisher CA, Malik N, Mitson MJ, Steensma DP et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat 2008; 29: 796–802.
Wada T, Kubota T, Fukushima Y, Saitoh S . Molecular genetic study of Japanese patients with X-linked alpha-thalassemia/mental retardation syndrome (ATR-X). Am J Med Genet 2000; 94: 242–248.
Badens C, Lacoste C, Philip N, Martini N, Courrier S, Giuliano F et al. Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin Genet 2006; 70: 57–62.
Picketts DJ, Tastan AO, Higgs DR, Gibbons RJ . Comparison of the human and murine ATRX gene identifies highly conserved, functionally important domains. Mamm Genome 1998; 9: 400–403.
Gibbons R . Alpha thalassaemia-mental retardation, X linked. Orphanet J Rare Dis 2006; 1: 15.
Ausió J, Levin DB, De Amorim GV, Bakker S, Macleod PM . Syndromes of disordered chromatin remodeling. Clin Genet 2003; 64: 83–95.
Horesh N, Pery R, Amiel I, Shwaartz C, Speter C, Guranda L et al. Volvulus and bowel obstruction in ATR-X syndrome-clinical report and review of literature. Am J Med Genet A 2015; 167A: 2777–2779.
Data Citations
Hasegawa, Tomonobu HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.1345
Acknowledgements
We thank the patient and their families for participation in this study. We also thank Kazue Kinoshita for the technical assistance. This work was supported by a Grant from the Japan Society for the Promotion of Science (16K10007 to MT) and Takeda Science Foundation (to MT), The Japanese Society for Pediatric Endocrinology Future Development Grant supported by Novo Nordisk Pharma (to MT), the Foundation for Growth Science, Japan (to MT), and the grant from the Ministry of Health, Labour and Welfare, Japan (Jitsuyoka Nanbyo-Ippan-014 to TH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Takagi, M., Yagi, H., Fukuzawa, R. et al. Syndromic disorder of sex development due to a novel hemizygous mutation in the carboxyl-terminal domain of ATRX. Hum Genome Var 4, 17012 (2017). https://doi.org/10.1038/hgv.2017.12
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.12
