
Abstract
The human noggin (NOG) gene is responsible for a broad spectrum of clinical manifestations of NOG-related symphalangism spectrum disorder (NOG-SSD), which include proximal symphalangism, multiple synostoses, stapes ankylosis with broad thumbs (SABTT), tarsal–carpal coalition syndrome, and brachydactyly type B2. Some of these disorders exhibit phenotypes associated with congenital stapes ankylosis. In the present study, we describe a Japanese pedigree with dactylosymphysis and conductive hearing loss due to congenital stapes ankylosis. The range of motion in her elbow joint was also restricted. The family showed multiple clinical features and was diagnosed with SABTT. Sanger sequencing analysis of the NOG gene in the family members revealed a novel heterozygous nonsense mutation (c.397A>T; p.K133*). In the family, the prevalence of dactylosymphysis and hyperopia was 100% while that of stapes ankylosis was less than 100%. Stapes surgery using a CO2 laser led to a significant improvement of the conductive hearing loss. This novel mutation expands our understanding of NOG-SSD from clinical and genetic perspectives.
Similar content being viewed by others

Introduction
The human noggin (NOG) gene consists of a single exon and encodes a secreted protein that is critical for normal bone and joint development.1 Noggin binds to bone morphogenetic protein (BMP) of the transforming growth factor-β superfamily and prevents its binding to the cognate receptor.1,2 This interaction affects a number of developmental processes such as morphogenesis and body patterning,1,3 middle ear formation,4,5 and apoptosis in digital and interdigital regions.4,6,7 Mutations in the NOG gene result in aberrant functioning of the noggin protein, which is linked to various autosomal dominant syndromes characterized by proximal symphalangism (SYM1: MIM #185800),8 multiple synostosis syndrome (SYNS1: MIM#186500),8 tarsal–carpal coalition syndrome (TCC: MIM#186570),9,10 brachydactyly type B2 (BDB2: MIM#611377),11 and stapes ankylosis with broad thumb and toes (SABTT: MIM#184460) (i.e., Teunissen-Cremers syndrome).12–14 Precise diagnosis is complicated by the overlapping clinical features of these syndromes. Given the variable phenotypic manifestations within and among families with the same mutations, the term NOG-related symphalangism spectrum disorder (NOG-SSD) has been proposed.15
In the present study, we describe a novel nonsense mutation in the NOG gene causing familial NOG-SSD and report the associated clinical and molecular findings as well as the results of surgery for conductive hearing loss.
Materials and methods
Patients
Medical history including hearing loss, symphalangism, dactylosymphysis, brachydactyly, and hyperopia as well as results of a clinical examination were obtained for four members of a Japanese family, three of whom were affected (proband, father, and grandmother), and one who was unaffected (mother). Auditory function was assessed by pure tone audiometry, tympanometry, and the stapedius reflex test. High-resolution computed tomography scans were carried out to identify any middle and inner ear abnormalities, and X-rays images of the hand were obtained to identify any fusion of the bones. Stapes surgery was performed in the proband to restore hearing. The father had undergone stapes surgery in the right ear at a different hospital in his childhood.
Study participants and the parents of the child provided written, informed consent. The research protocol was approved by the Ethical Review Committee of Sapporo Medical University, Japan.
Genetic analysis
Genomic DNA was extracted from blood samples using the Gentra Puregene Blood kit (Qiagen, Hamburg, Germany). PCR primers specific for the NOG exon (GenBank NG_011958.1) and the amplification program were as previously reported.16 Sanger sequencing data were analyzed using SeqScape software v.2.6 (Applied Biosystems, Foster City, CA, USA) and DNASIS Pro (Hitachisoft, Tokyo, Japan). The variant allele frequency was evaluated using the dbSNP 146 public database (http://www.ncbi.nlm.nih.gov/snp/), 1000 Genome Browser (http://browser.1000genomes.org/index.html), Human Genetic Variation Database (HGVD) (http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html), NHLBI Exome Sequencing Project (ESP6500) (http://evs.gs.washington.edu/EVS/), and Exome Aggregation Consortium v.0.3 (ExAC) (http://exac.broadinstitute.org). The Human Gene Mutation Database (HGMD) v.2015.4 (BIOBASE, Beverly, MA, USA) was searched to determine whether the variant had been previously reported as being associated with diseases.
Results
Clinical features of the family
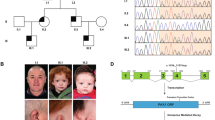
The family had five affected individuals (Figure 1). The proband (IV: 1) was a 6-year-old girl of non-consanguineous Japanese parents. She was referred to our hospital at the age of 5 years because of bilateral hearing loss that had starting in early childhood. Physical and X-ray examinations of the hands showed symphalangism and short intermediate phalanges (brachydactyly) in both fifth fingers (Figure 2a). The range of motion in her elbow joint was restricted, and she was unable to touch her shoulders with her hands (Figure 2f).

Pedigree of a family with NOG-SSD. The family included five affected individuals (I: 2, II: 2, II: 3, III: 3, and IV: 1 (proband)). Arrows indicate subjects who participated in the genetic analysis.

Photographs of the hands of the proband (a), her father (b), and her grandmother (c). Arrows indicate symphalangism and arrowheads indicate brachydactyly. (d–f) Photograph illustrating the restricted range of motion of the elbow joint in the affected individuals and their inability to touch their shoulders with their hands.
She had undergone surgical treatment for dactylosymphysis in the second and third toes of both feet in her early childhood. An ophthalmologic examination revealed hyperopia. She had experienced bilateral progressive hearing loss from early childhood, and pure tone audiometry at age 6 showed bilateral conductive hearing loss (Figure 3a). At this time, she also underwent stapedotomy using a Teflon piston, which detected ankylosis of the stapes footplate with hypertrophy of the anterior and posterior crus; the footplate was also distant from the facial nerve (Figure 4). The patient’s postoperative hearing threshold improved to 25 dB in the operated ear, and her hearing level has remained stable for more than 3 years since the surgery (Figure 3b).

(a) Preoperative pure tone audiometry of the proband showing bilateral conductive hearing loss. (b) Postoperative pure tone audiometry demonstrating improvement in hearing levels in the operated ear. (c) Audiograms from III: 3 showed conductive hearing loss in the left ear, while the right ear treated by stapes surgery showed an improvement in hearing threshold. (d) Audiograms from II: 2 did not reveal conductive hearing loss.

Operative findings in the proband showing ankylosis of the stapes footplate with hypertrophy of the anterior and posterior crus. The facial nerve was distant from the footplate.
The proband’s father (III: 3), who underwent right stapedotomy at 13 years old at another hospital, had bilateral hearing loss since early childhood, and pure tone audiometry showed conductive hearing loss on the left side and an improvement of hearing level in the operated ear (Figure 3c). His hearing condition had not worsened for 15 years. Physical examination of his hands showed brachydactyly in both fifth fingers (Figure 2b), and he could not touch his shoulders with his hands due to a restricted range of motion in his elbow joints (Figure 2d). He had undergone surgical treatment in his early childhood for dactylosymphysis in the second and third toes of both feet. An ophthalmologic examination revealed hyperopia.
The proband’s grandmother (II: 2) did not show conductive hearing loss (Figure 3d); however, she had also had surgery during childhood for dactylosymphysis in the second and third toes of the both feet. Physical examination of her hands did not reveal brachydactyly (Figure 2c). As in the case of the other two patients, she was unable to touch her shoulders with her hands due to restricted range of motion of the elbow joint (Figure 2e). An ophthalmologic examination showed hyperopia.
Pilonidal cysts were found in the proband (IV: 1) and her father (III: 3).
Genetic analysis
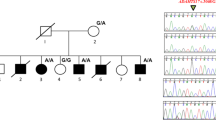
A genetic analysis detected a heterozygous c.397A>T (p.K133*) variant of the NOG gene in the proband (IV: 1) as well as in II-2 and III-3 (Figure 5), which has not been previously reported according to the HGMD and is not registered in other databases such as dbSNP, 1000 Genome Browser, HGVD, ESP6500, or ExAC. Given that other nonsense mutations such as p.Q110* (rs104894614)14 and p.L129* (rs104894613)17 have been reported to be pathogenic, the p.K133* variant is presumed to produce a truncated noggin protein (132 of 232 amino acid residues) with disrupted function.

Partial electropherograms of NOG from a patient with c. 397A>T (p.K133*) mutation (left) and a control subject with normal hearing (right). The mutated nucleotide is indicated by an arrow.
Discussion
The present study identified a novel nonsense mutation in the NOG gene in a family with NOG-SSD. The clinical features included proximal symphalangism in one of the fingers, dactylosymphysis of the toes, brachydactyly, pilonidal cyst, hyperopia, and conductive hearing loss as a result of stapes ankylosis. The most common phenotypes in the family were dactylosymphysis (5/5), hyperopia (5/5), and hearing loss (4/5).
Heterozygous NOG mutations have been identified in several syndromes including SYM1,8 SYNS1,8 TCC,9,10 BDB2,11 and SABTT.12–14 To date, a total of 45 human variations in NOG have been reported; the term NOG-SSD was put forth to describe these syndromes,15,17 which exhibit shared but also some distinct clinical features. In our patients, the prevalence of dactylosymphysis and hyperopia was 100% while that of stapes ankylosis was less than 100%.
Mutations reported in the literature to date are shown in Table 1. NOG gene mutations including frameshift, missense, and nonsense mutations as well as deletions and insertions have been previously identified in patients with NOG-SSD. NOG gene mutations are mainly dominant; however, de novo mutations have also been reported in sporadic cases.8,18 Therefore, genetic investigations are sometimes needed to clarify the pathogenesis of conductive hearing loss due to stapes ankylosis with stiffness of the proximal interphalangeal joints in patients with no familial history. NOG gene mutations are autosomal dominant, and is presumed to be manifested as either haploinsufficiency—which can lead to an aberrant gradient during development—or may have a dominant-negative effect due to the defective protein.19 The NOG gene has a critical role in joint formation and bone development, and mutations in noggin compromise the folding stability of the protein and cause defective binding to BMP.6,20 Noggin-mediated inhibition of BMP signaling is regulated by a two-step process:21 noggin binds to BMP and prevents its binding to the BMP receptor, with the complex binding instead to heparin sulfate proteoglycan, a major cell surface and extracellular matrix proteoglycan. Sulfate induces the release of the noggin–BMP complex at the cell surface, increasing the accessibility of BMP to its receptor and thereby activating BMP signaling. A docking simulation of noggin to heparin analog and estimation of the change in interaction with p.R136C mutation demonstrated that the positively charged R136 in the heparin-binding site is required for retention of the noggin–BMP complex by negatively charged heparin sulfate proteoglycan at the plasma membrane.16 The altered binding of mutant noggin and heparin sulfate proteoglycan may lead to hyperactivation of BMP signaling, ultimately leading to ankylosis of the joints and stapes.16
Stapes surgery for conductive hearing loss due to NOG mutations leads to an improvement in hearing for most patients,10,19,22 as confirmed in the present study. However, it is necessary to exercise caution when performing stapes surgery for this syndrome due to the risk of bony reclosure of the oval window after surgery. It was previously reported that the hearing level of patients who underwent stapes surgery deteriorated during the follow-up period for this reason, which resulted in a dislocated piston.18,23,24 Therefore, partial or total stapedectomy has been proposed as an alternative procedure to prevent reclosure of the oval window.7,23 In the present case (IV: 1), we performed stapedotomy using a CO2 laser. There have been no reports to date of CO2 laser-assisted stapedotomy for treatment of stapes ankylosis due to NOG mutations; therefore, the surgical outcome must be carefully assessed after long-term follow-up.
In conclusion, we identified a novel nonsense mutation in the NOG gene (p.K133*) in a NOG-SSD family. NOG gene mutations lead to aberrant functioning of the noggin protein, giving rise to a large spectrum of clinical features. Our patients exhibited a phenotype that included proximal symphalangism, dactylosymphysis, brachydactyly of the toes, pilonidal cyst, hyperopia, and conductive hearing loss. Stapes surgery for conductive hearing loss is a good therapeutic option; however, patients should be carefully monitored over the long term. This novel mutation and clinical manifestations contribute to a better understanding of NOG-SSD.
References
Zimmerman LB, De Jesus-Escobar JM, Harland RM . The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein. Cell 1996; 86: 599–606.
Smith WC, Harland RM . Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell 1992; 70: 829–840.
Hogan BL . Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 1996; 10: 1580–1594.
Declau F, Van den Ende J, Baten E, Mattelaer P . Stapes ankylosis in a family with a novel NOG mutation: otologic features of the facioaudiosymphalangism syndrome. Otol Neurotol 2005; 26: 934–940.
Hwang CH, Wu DK . Noggin heterozygous mice: an animal model for congenital conductive hearing loss in humans. Hum Mol Genet 2008; 17: 844–853.
Brunet LJ, McMahon JA, McMahon AP, Harland RM . Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 1998; 280: 1455–1457.
Weekamp HH, Kremer H, Hoefsloot LH, Kuijpers-Jagtman AM, Cruysberg JR, Cremers CW . Teunissen-Cremers syndrome: a clinical, surgical, and genetic report. Otol Neurotol 2005; 26: 38–51.
Gong Y, Krakow D, Marcelino J, Wilkin D, Chitayat D, Babul-Hirji R et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 1999; 21: 302–304.
Drawbert JP, Stevens DB, Cadle RG, Hall BD . Tarsal and carpal coalition and symphalangism of the Fuhrmann type. Report of a family. J Bone Joint Surg Am 1985; 67: 884–889.
Gregersen HN, Petersen GB . Congenital malformation of the feet with low body height. A new syndrome, caused by an autosomal dominant gene. Clin Genet 1977; 12: 255–262.
Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet 2007; 81: 388–396.
Teunissen B, Cremers WR . An autosomal dominant inherited syndrome with congenital stapes ankylosis. Laryngoscope 1990; 100: 380–384.
Merino R, Ganan Y, Macias D, Economides AN, Sampath KT, Hurle JM . Morphogenesis of digits in the avian limb is controlled by FGFs, TGF betas, and noggin through BMP signaling. Dev Biol 1998; 200: 35–45.
Brown DJ, Kim TB, Petty EM, Downs CA, Martin DM, Strouse PJ et al. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet 2002; 71: 618–624.
Potti TA, Petty EM, Lesperance MM . A comprehensive review of reported heritable noggin-associated syndromes and proposed clinical utility of one broadly inclusive diagnostic term: NOG-related-symphalangism spectrum disorder (NOG-SSD). Hum Mutat 2011; 32: 877–886.
Masuda S, Namba K, Mutai H, Usui S, Miyanaga Y, Kaneko H et al. A mutation in the heparin-binding site of noggin as a novel mechanism of proximal symphalangism and conductive hearing loss. Biochem Biophys Res Commun 2014; 447: 496–502.
Usami S, Abe S, Nishio S, Sakurai Y, Kojima H, Tono T et al. Mutations in the NOG gene are commonly found in congenital stapes ankylosis with symphalangism, but not in otosclerosis. Clin Genet 2012; 82: 514–520.
Takahashi T, Takahashi I, Komatsu M, Sawaishi Y, Higashi K, Nishimura G et al. Mutations of the NOG gene in individuals with proximal symphalangism and multiple synostosis syndrome. Clin Genet 2001; 60: 447–451.
Thomeer HG, Admiraal RJ, Hoefsloot L, Kunst HP, Cremers CW . Proximal symphalangism, hyperopia, conductive hearing impairment, and the NOG gene: 2 new mutations. Otol Neurotol 2011; 32: 632–638.
Groppe J, Greenwald J, Wiater E et al. Structural basis of BMP signalling inhibition by the cysteine knot protein Noggin. Nature 2002; 420: 636–642.
Viviano BL, Paine-Saunders S, Gasiunas N, Gallagher J, Saunders S . Domain specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J Biol Chem 2004; 279: 5604–5611.
Ganaha A, Kaname T, Akazawa Y, Higa T, Shinjou A, Naritomi K et al. Identification of two novel mutations in the NOG gene associated with congenital stapes ankylosis and symphalangism. J Hum Genet 2015; 60: 27–34.
Ensink RJ, Sleeckx JP, Cremers CW . Proximal symphalangism and congenital conductive hearing loss: otologic aspects. Am J Otol 1999; 20: 344–349.
Brown DJ, Kim TB, Petty EM, Downs CA, Martin DM, Strouse PJ et al. Characterization of a stapes ankylosis family with a NOG mutation. Otol Neurotol 2003; 24: 210–215.
Hirshoren N, Gross M, Banin E, Sosna J, Bargal R, Raas-Rothschild A . P35S mutation in the NOG gene associated with Teunissen-Cremers syndrome and features of multiple NOG joint-fusion syndromes. Eur J Med Genet 2008; 51: 351–357.
Mangino M, Flex E, Digilio MC, Giannotti A, Dallapiccola B . Identification of a novel NOG gene mutation (P35S) in an Italian family with symphalangism. Hum Mutat 2002; 19: 308.
Dixon ME, Armstrong P, Stevens DB, Bamshad M . Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet Med 2001; 3: 349–353.
Debeer P, Fryns JP, Devriendt K, Baten E, Huysmans C, Van de Ven WJ . A novel NOG mutation Pro37Arg in a family with tarsal and carpal synostoses. Am J Med Genet A 2004; 128: 439–440.
Debeer P, Huysmans C, Van de Ven WJ, Fryns JP, Devriendt K . Carpal and tarsal synostoses and transverse reduction defects of the toes in two brothers heterozygous for a double de novo NOGGIN mutation. Am J Med Genet A 2005; 143: 318–320.
Oxley CD, Rashid R, Goudie DR, Stranks G, Baty DU, Lam W et al. Growth and skeletal development in families with NOGGIN gene mutations. Horm Res 2008; 69: 221–226.
Pang X, Wang Z, Chai Y, Chen H, Li L, Sun L et al. A novel missense mutation of NOG interferes with the dimerization of NOG and causes proximal symphalangism syndrome in a Chinese family. Ann Otol Rhinol Laryngol 2015; 124: 745–751.
Liu F, Huang Y, Liu L et al. Identification of a novel NOG mutation in a Chinese family with proximal symphalangism. Clin Chim Acta 2014; 15: 129–133.
Dawson K, Seeman P, Sebald E, King L, Edwards M, Williams J 3rd et al. GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet 2006; 78: 708–712.
van den Ende JJ, Mattelaer P, Declau F, Vanhoenacker F, Claes J, Van Hul E et al. The facio-audio-symphalangism syndrome in a four generation family with a nonsense mutation in the NOG-gene. Clin Dysmorphol 2005; 14: 73–80.
Emery SB, Meyer A, Miller L, Lesperance MM . Otosclerosis or congenital stapes ankylosis? The diagnostic role of genetic analysis. Otol Neurotol 2009; 30: 1204–1208.
Ishino T, Takeno S, Hirakawa K . Novel NOG mutation in Japanese patients with stapes ankylosis with broad thumbs and toes. Eur J Med Genet 2015; 58: 427–432.
Rudnik-Schöneborn S, Takahashi T, Busse S, Schmidt T, Senderek J, Eggermann T et al. Facioaudiosymphalangism syndrome and growth acceleration associated with a heterozygous NOG mutation. Am J Med Genet A 2010; 152: 1540–1544.
Shimizu R, Mitsui N, Mori Y, Cho S, Yamamori S, Osawa M et al. Cryptic 17q22 deletion in a boy with a t(10;17)(p15.3;q22) translocation, multiple synostosis syndrome 1, and hypogonadotropic hypogonadism. Am J Med Genet A 2008; 146: 1458–1461.
Pang X, Luo H, Chai Y, Wang X, Sun L, He L et al. A 1.6-Mb microdeletion in chromosome 17q22 leads to NOG-related symphalangism spectrum disorder without intellectual disability. PLoS One 2015; 10: e0120816.
Acknowledgements
This research was partially supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grant no. 25861575).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Takano, K., Ogasawara, N., Matsunaga, T. et al. A novel nonsense mutation in the NOG gene causes familial NOG-related symphalangism spectrum disorder. Hum Genome Var 3, 16023 (2016). https://doi.org/10.1038/hgv.2016.23
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.23
This article is cited by
-
The roles and regulatory mechanisms of TGF-β and BMP signaling in bone and cartilage development, homeostasis and disease
Cell Research (2024)
-
Identification of a novel nonsense NOG mutation in a patient with stapes ankylosis and symphalangism spectrum disorder
Human Genome Variation (2023)
-
A defect in the NOG gene increases susceptibility to spontaneous superficial chronic corneal epithelial defects (SCCED) in boxer dogs
BMC Veterinary Research (2021)
-
Genetic and clinical phenotypic analysis of familial stapes sclerosis caused by an NOG mutation
BMC Medical Genomics (2020)
-
Novel NOG (p.P42S) mutation causes proximal symphalangism in a four-generation Chinese family
BMC Medical Genetics (2019)
