
Abstract
Glaucoma is the second leading cause of blindness, affecting ~65 million people worldwide. We identified and ascertained a large cohort of inbred families with multiple individuals manifesting cardinal symptoms of primary congenital glaucoma (PCG) to investigate the etiology of the disease at a molecular level. Ophthalmic examinations, including slit-lamp microscopy and applanation tonometry, were performed to characterize the causal phenotype and confirm that affected individuals fulfilled the diagnostic criteria for PCG. Subsequently, exclusion analysis was completed with fluorescently labeled short tandem repeat markers, followed by Sanger sequencing to identify pathogenic variants. Exclusion analysis suggested linkage to the CYP1B1 locus, with positive two-point logarithm of odds scores in 23 families, while Sanger sequencing identified a total of 11 variants, including two novel mutations, in 23 families. All mutations segregated with the disease phenotype in their respective families. These included the following seven missense mutations: p.Y81N, p.E229K, p.R368H, p.R390H, p.W434R, p.R444Q and p.R469W, as well as one nonsense mutation, p.Q37*, and three frameshift mutations, p.W246Lfs81*, p.T404Sfs30* and p.P442Qfs15*. In conclusion, we identified a total of 11 mutations, reconfirming the genetic heterogeneity of CYP1B1 in the pathogenesis of PCG. To the best of our knowledge, this is the largest study investigating the contribution of CYP1B1 to the pathogenesis of PCG in the Pakistani population.
Similar content being viewed by others

Glaucoma is the second leading cause of blindness, affecting ~65 million people worldwide.1 It causes irreversible visual field defects, eventually resulting in complete blindness.2,3 Glaucoma encompasses a range of ocular dystrophies that each exhibit optic disc neuropathy, which is specifically characterized by progressive loss of retinal ganglion cells and optic nerve atrophy.4,5 The disease is generally classified by age of onset, anatomy of the anterior chamber and etiology. These characteristics define the following three broad categories of disease: open-angle glaucoma, closed-angle glaucoma and primary congenital glaucoma.6
The focus of our study, primary congenital glaucoma (PCG), is characterized by developmental defects in the anterior chamber that obstruct the normal flow of aqueous fluid in the eye. Blockage and subsequent buildup of fluid increases intraocular pressure (IOP) and damages the optic nerve.7–10 Clinical indications of PCG include elevated IOP, buphthalmos (globe enlargement), photophobia, corneal enlargement, epiphora, Haab’s striae (Descemet’s membrane ruptures), blepharospasm and optic nerve damage.7,8 PCG occurs in approximately 1 in 10,000 live births, but the incidence varies widely between populations.6
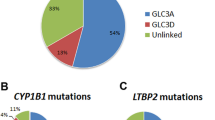
PCG is a genetically heterogeneous disorder that occurs both sporadically and in families and is inherited as an autosomal recessive trait. Linkage analysis has identified the following four loci: GLC3A (2p22-p21), GLC3B (1p36.2-36.1), GLC3C (14q24.3) and GLC3D (14q24.2-24.3).11–14 GLC3A and GLC3D have been cloned, with mutations in cytochrome P450, subfamily B, polypeptide 1 (CYP1B1) and latent transforming growth factor β binding protein 2 (LTBP2) reported in populations of multiple ethnicities.15,16 We previously reported four causal mutations identified in large consanguineous families of Pakistani origin.14
In an ongoing effort to investigate the genetic basis of PCG and identify novel disease loci and/or causal mutations responsible for the disease phenotype, we have recruited a large cohort of consanguineous families, each including at least one affected individual from a consanguineous mating. Approval for the study was granted by the Institutional Review Boards (IRBs) of the National Centre of Excellence in Molecular Biology (Lahore, Pakistan), the National Eye Institute (Bethesda, MD, USA) and Johns Hopkins University (Baltimore, MD, USA).
Individuals who participated in this study signed an informed written consent, which was approved by each IRB and adheres to the Declaration of Helsinki. Any available medical records were used to compile medical histories for the study participants, but much of the information was provided in interviews with the families. The majority of the enrolled families live in remote areas without access to ophthalmic clinics. Therefore, the precise age of onset of glaucoma or the age at first diagnosis is not well documented. The parents of the affected children, as well as family elders, reported that reduced or abnormal vision was evident in the first year of the children’s lives. The consistently early onset of symptoms suggests congenital ocular dystrophy. Common symptoms of the affected individuals were increased IOP, increased corneal diameter and visual acuity that was reduced to hand movement and/or light perception (Table 1).
All study participants provided a blood sample of ~10 ml that was collected in a tube containing 400 μl of 0.5 mol/l EDTA. Genomic DNA was extracted from white blood cells as previously described.17 Exclusion analysis, two-point logarithm of odds (LOD) score calculations, and bi-directional Sanger sequencing were completed as previously described, along with the sequences of the primer pairs used for Sanger sequencing.14
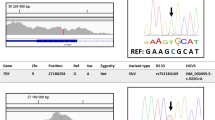
A total of 23 familial cases localized to chromosome 2p21 (Supplementary Figures 1–23): PKGL001, PKGL014, PKGL028, PKGL032, PKGL040, PKGL046, PKGL047, PKGL050, PKGL051, PKGL058, PKGL060, PKGL065, PKGL066, PKGL067, PKGL068, PKGL069, PKGL070, PKGL071, PKGL072, PKGL073, PKGL077, PKGL079 and PKGL082. Alleles of chromosome 2p21 markers provided evidence of linkage to the GLC3A locus, with positive LOD scores (Supplementary Tables 1–23). Subsequently, we sequenced all coding exons, exon–intron boundaries, and the 5′ and 3′ UTR regions of CYP1B1. In 23 PCG families, we identified 11 homozygous variations, including seven missense mutations, one nonsense mutation and three frameshift mutations (Table 2).
We identified a homozygous variation, c.1405C>T (p.R469W), in both PKGL001 and PKGL028. Similarly, we identified a homozygous variation, c.1169G>A (p.R390H), in PKGL040, PKGL046, PKGL060, PKGL065, PKGL066, PKGL067, PKGL069, PKGL070, PKGL071, PKGL073, PKGL077, PKGL079 and PKGL082. Moreover, we identified the homozygous variations c.1300T>C (p.W434R), c.685G>A (p.E229K), c.1331G>A (p.R444Q), c.241T>A (p.Y81N), c.1103G>A (p.R368H), and c.109C>T (p.Q37*) in PKGL014, PKGL047, PKGL050, PKGL051, PKGL058 and PKGL072, respectively (Table 2).
We further identified a homozygous 1 bp insertion, c.736_737insT (p.W246Lfs81*), and a homozygous 1 bp deletion, c.1325delC (p.P442Qfs15*), in PKGL047 and PKGL068, respectively (Table 2 and Supplementary Figure 24a,b), and a 10-bp homozygous duplication, c.1200_1209dupTCATGCCACC (p.T404Sfs30*), in PKGL032 (Table 2). Each of these mutations results in a frameshift and eventually leads to premature termination of CYP1B1. Among these variants, seven missense mutations, a frameshift mutation, and a nonsense mutation have been previously reported, whereas p.W246Lfs81* and p.P442Qfs15* are novel. All the above-mentioned pathogenic variations co-segregated with the disease phenotype in their respective families and were absent in 192 ethnically matched control chromosomes.
Next, we examined the evolutionary conservation of the mutated amino acids by aligning the protein sequences of CYP1B1 orthologs. In parallel, we investigated the possible effect of amino acid substitution on the structure of CYP1B1 with SIFT (http://sift.jcvi.org), Condel (http://bg.upf.edu/fannsdb/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml). Evolutionary conservation of the substituted amino acids of all seven missense variants show that not only these amino acids but also the amino acids in the immediate vicinity are highly conserved among other primates, placental mammals and vertebrates (Supplementary Figure 24c). SIFT and PolyPhen-2 analyses showed that all 11 pathogenic variants were damaging to the function and enzymatic activity of CYP1B1.
CYP1B1, a member of the cytochrome P450 superfamily, is the largest known enzyme of the human cytochrome P450 pathway that is primarily expressed in the trabecular meshwork, iris, retina and ciliary body. Membrane-bound CYP1B1 has a molecular structure consisting of a 53-residue membrane-spanning domain located at the NH2 (amino) terminus of the molecule, followed by a 10-residue proline-rich ‘hinge’ region, which allows flexibility between the membrane-spanning and cytoplasmic domains of the protein molecule.18 The COOH (carboxy) terminus of CYP1B1 is highly conserved, consisting of conserved core structures composed of a J-helix, a K-helix and a heme-binding region.19,20 Mutations in CYP1B1, either at the NH2 or the COOH terminus, are expected to interfere with the basic properties of the CYP1B1 molecule, such as its ability to bind heme or to adopt its regular conformation, resulting in impaired enzymatic function.20,21 The pathogenesis of CYP1B1 in PCG is still not fully understood, but it is thought to be involved in metabolic pathways related to ocular differentiation (anterior segment and trabecular meshwork formation). A study conducted recently on CYP1B1-deficient mice showed that CYP1B1 deficiency resulted in increased oxidative stress and structural defects in the trabecular meshwork in the early lives of mice.22
To date, more than 150 mutations in CYP1B1 have been associated with PCG, accounting for a significant fraction of the genetic load of familial and sporadic cases of PCG. We found one mutation, R390H, in 13 of the 23 familial cases investigated in this study, accounting for more than half of the families linked to CYP1B1, which is consistent with previously published estimates. The R390H allele is the most frequently occurring mutation identified in Chinese, Iranian, Indian and Pakistani populations affected by PCG, while only a small fraction of Caucasians harbor the R390H allele.23–27
We further investigated the origin of this mutation by constructing a haplotype exploiting the following six previously reported CYP1B1 SNPs: rs2617266, rs10012, rs1056827, rs1056836, rs1056837 and rs1800440. All 13 families harboring the R390H allele shared a common haplotype, C–C–G–C–C–G (data not shown). The same haplotype was recently reported in five families of Pakistani origin that harbored the R390H allele.27
Interestingly, the W434R allele identified in PKGL014 has previously been implicated in primary open-angle glaucoma (POAG), where a single heterozygous carrier of Indian descent was reported to have POAG.28 However, no clinical details of the patient were included in the report; therefore, we cannot compare the severity of the causal phenotype of the heterozygous carrier reported to have POAG with that of the affected individuals in PKGL014 who are homozygous for the W434R allele. Surprisingly, the heterozygous carriers of the W434R allele in PKGL014 are phenotypically normal and do not show any signs or clinical symptoms of POAG or any other ocular dystrophy.
In conclusion, we report a broad spectrum of mutations identified in CYP1B1 that are responsible for PCG in consanguineous families of Pakistani origin, reaffirming the role of CYP1B1 in the pathogenesis of PCG. To the best of our knowledge, this is the largest study investigating the contributions of CYP1B1 causal mutations associated with PCG in the Pakistani population, which will add to our understanding of the genetic basis of PCG.
References
References
Pascolini D, Mariotti SP . Global estimates of visual impairment: 2010. Br J Ophthalmol 2012; 96: 614–618.
Ray K, Mukhopadhyay A, Acharya M . Recent advances in molecular genetics of glaucoma. Mol Cell Biochem 2003; 253: 223–231.
Quigley HA, Broman AT . The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 2006; 90: 262–267.
Libby RT, Gould DB, Anderson MG, John SW . Complex genetics of glaucoma susceptibility. Annu Rev Genomics Hum Genet 2005; 6: 15–44.
Quigley HA . Open-angle glaucoma. N Engl J Med 1993; 328: 1097–1106.
Sarfarazi M, Stoilov I, Schenkman JB . Genetics and biochemistry of primary congenital glaucoma. Ophthalmol Clin North Am 2003; 16: 543–554, vi.
Barkan O . Pathogenesis of congenital glaucoma: gonioscopic and anatomic observation of the angle of the anterior chamber in the normal eye and in congenital glaucoma. Am J Ophthalmol 1955; 40: 1–11.
Maumenee AE . The pathogenesis of congenital glaucoma: a new theory. Trans Am Ophthalmol Soc 1958; 56: 507–570.
Kupfer C, Kaiser-Kupfer MI . Observations on the development of the anterior chamber angle with reference to the pathogenesis of congenital glaucomas. Am J Ophthalmol 1979; 88: 424–426.
Anderson DR . The development of the trabecular meshwork and its abnormality in primary infantile glaucoma. Trans Am Ophthalmol Soc 1981; 79: 458–485.
Sarfarazi M, Akarsu AN, Hossain A, Turacli ME, Aktan SG, Barsoum-Homsy M et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics 1995; 30: 171–177.
Akarsu AN, Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, Sayli BS et al. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum Mol Genet 1996; 5: 1199–1203.
Stoilov IR, Costa VP, Vasconcellos JP, Melo MB, Betinjane AJ, Carani JC et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci 2002; 43: 1820–1827.
Firasat S, Riazuddin SA, Khan SN, Riazuddin S . Novel CYP1B1 mutations in consanguineous Pakistani families with primary congenital glaucoma. Mol Vis 2008; 14: 2002–2009.
Stoilov I, Akarsu AN, Sarfarazi M . Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet 1997; 6: 641–647.
Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet 2009; 84: 664–671.
Khan SY, Vasanth S, Kabir F, Gottsch JD, Khan AO, Chaerkady R et al. FOXE3 contributes to Peters anomaly through transcriptional regulation of an autophagy-associated protein termed DNAJB1. Nat Commun 2016; 7: 10953.
Yamazaki S, Sato K, Suhara K, Sakaguchi M, Mihara K, Omura T . Importance of the proline-rich region following signal-anchor sequence in the formation of correct conformation of microsomal cytochrome P-450s. J Biochem 1993; 114: 652–657.
Swindell EC, Eichele G . Retinoid metabolizing enzymes in development. Biofactors 1999; 10: 85–89.
Chen H, Howald WN, Juchau MR . Biosynthesis of all-trans-retinoic acid from all-trans-retinol: catalysis of all-trans-retinol oxidation by human P-450 cytochromes. Drug Metab Dispos 2000; 28: 315–322.
Sarfarazi M, Stoilov I . Molecular genetics of primary congenital glaucoma. Eye (Lond) 2000; 14 (Pt 3B): 422–428.
Zhao Y, Sorenson CM, Sheibani N . Cytochrome P450 1B1 and primary congenital glaucoma. J Ophthalmic Vis Res 2015; 10: 60–67.
Yang M, Guo X, Liu X, Shen H, Jia X, Xiao X et al. Investigation of CYP1B1 mutations in Chinese patients with primary congenital glaucoma. Mol Vis 2009; 15: 432–437.
Li N, Zhou Y, Du L, Wei M, Chen X . Overview of cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res 2011; 93: 572–579.
Chitsazian F, Tusi BK, Elahi E, Saroei HA, Sanati MH, Yazdani S et al. CYP1B1 mutation profile of Iranian primary congenital glaucoma patients and associated haplotypes. J Mol Diagn 2007; 9: 382–393.
Tanwar M, Dada T, Sihota R, Das TK, Yadav U, Dada R . Mutation spectrum of CYP1B1 in North Indian congenital glaucoma patients. Mol Vis 2009; 15: 1200–1209.
Sheikh SA, Waryah AM, Narsani AK, Shaikh H, Gilal IA, Shah K et al. Mutational spectrum of the CYP1B1 gene in Pakistani patients with primary congenital glaucoma: novel variants and genotype-phenotype correlations. Mol Vis 2014; 20: 991–1001.
Chakrabarti S, Devi KR, Komatireddy S, Kaur K, Parikh RS, Mandal AK et al. Glaucoma-associated CYP1B1 mutations share similar haplotype backgrounds in POAG and PACG phenotypes. Invest Ophthalmol Vis Sci 2007; 48: 5439–5444.
Data Citations
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.805
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.808
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.811
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.814
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.817
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.820
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.823
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.826
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.829
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.832
Riazuddin, S Amer HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.835
Acknowledgements
We are thankful to all the participants for their time and cooperation and for their participation in this study. The study was funded in part by the National Academy of Sciences, Washington, DC, USA, and the Higher Education Commission, Islamabad, Pakistan.
Author information
Authors and Affiliations
Contributions
BR, BI, SF and SAR: conceived and designed the experiments; BR, BI and SF: recruited human subjects; SNK, TH, SR, JA and SAR: contributed reagents/materials/analysis tools; BR, BI, FK and SF: performed experiments; BR, BI, FK, SF, MAN, SNK, TH, SR, JA and SAR: analyzed the data; and BR, BI, FK, SR and SAR: contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Rauf, B., Irum, B., Kabir, F. et al. A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Hum Genome Var 3, 16021 (2016). https://doi.org/10.1038/hgv.2016.21
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.21
This article is cited by
-
Next-generation whole exome sequencing to delineate the genetic basis of primary congenital glaucoma
Scientific Reports (2022)
-
In silico analysis of five missense mutations in CYP1B1 gene in Pakistani families affected with primary congenital glaucoma
International Ophthalmology (2018)
