
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare, congenital disorder caused by heterozygous mutation of the bone morphogenetic protein type I receptor ACVR1. Various forms of atypical FOP have recently been identified, and a recurrent mutation, ACVR1 (p.Arg258Ser) was reported. We encountered a 17-year-old Japanese female patient with sporadic occurrence of FOP. At the age of 7 years, radiological examination revealed progressive heterotopic ossification and cervical spine malformations. Although great toe malformation was not observed, we diagnosed her as having FOP. Then, ACVR1 was analyzed and a recurrent mutation of p.Arg258Ser was identified. We noticed that there may be phenotypic differences between c.774G>T and c.774G>C, which lead to the same amino-acid change, p.Arg258Ser. Genotype–phenotype correlation was discussed with the review of the previous reports.
Similar content being viewed by others

Introduction
Fibrodysplasia ossificans progressiva (FOP, MIM#135100, http://www.omim.org/) is a rare hereditary disease caused by a heterozygous mutation in the type I activin receptor (ACVR1) gene that encodes a bone morphogenetic protein receptor. This disease leads to heterotopic ossification in the muscle tissue and the surrounding fascia, tendons and ligaments throughout the body.1 The allele exhibits variable expressivity and has complete penetrance. However, most patients have low reproductive fitness, and most cases are identified in spontaneous mutations of a gamete from either one of the healthy parents. There is no racial, geographic or gender predisposition; the worldwide prevalence is approximately one in every two million people.1,2 FOP is diagnosed when clinical symptoms and mutational analysis is confirmed. Ninety-seven percent of patients worldwide have classic FOP, which is defined by the presence of two classic clinical features: characteristic malformations of the great toes and onset of soft tissue flare-ups leading to progressive heterotopic ossification.
Because of the systemic heterotopic ossification, the pathological progression is associated with a restricted range of motion (ROM) affecting most joints. The ossification progression varies among individuals. Heterotopic ossification around the joints of the extremities causes extra-articular ankylosis, resulting in restricted activities of daily living, particularly difficulty in walking. Furthermore, some patients present with respiratory difficulties caused by heterotopic ossification in the spinal column and thorax. Progression of this condition reduces life expectancy.
As previously mentioned, although many reports have documented sporadic cases of FOP, familial cases due to autosomal dominant inheritance have also been reported. Linkage analysis of 32 sporadic and five familial FOP patients revealed a mutation (p.Arg206His) in the ACVR1 gene that was common to both sporadic and familial cases (classic FOP). To date, 11 point mutations have been identified in the ACVR1 gene.1–15 Among them, a recurrent mutation, NM_001105.4: p.Arg258Ser, was reported in the same kinase domain as the mutation reported in 2010 (p.Gly356Asp).14,15 Herein we report the results and analysis of the third patient with ACVR1 (p.Arg258Ser) caused by c.774G>T mutation.
Materials and methods
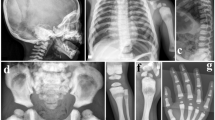
The patient was a 17-year old female. No other individuals in her family, including siblings, had FOP symptoms. The patient exhibited normal development and no notable restriction of ROM of her joints or trunk from birth until 7 years of age. At the age of 7 years, the patient fell from a swing and was examined by her local physician. X-ray imaging indicated fusion in her cervical vertebrae. Thereafter, the patient’s ROM in the shoulders and elbows gradually worsened. Biopsy of the cervical lesion was performed; however, no ossification was found. At the age of 14 years, the patient was examined by her local physician because of obvious body movement difficulties. Computed tomography scans revealed heterotopic ossification around the paraspinal muscles and bilateral shoulder and hip joints. The patient was referred to our institution at the age of 15 years and 2 months. At 17 years of age, the most recent findings were oral restriction (22 mm) and no evidence of scoliosis. In the upper extremities, restricted shoulder flexion (20/5°) and external rotation (40/5°) and ankyloses of the elbow joints were observed. Joint movements distal to the wrists were not affected. In the lower extremities, contracture of the right hip in external rotation and of the left hip in internal rotation resulted in a so-called ‘windblown deformity’. Restriction of ROM was also evident in the knees (85–110/70–110°) and ankle joint dorsiflexion (20/5°). The toes were generally short, without obvious great toe malformation. X-rays revealed no spinal deformity; however, there was heterotopic ossification of the bilateral paraspinal muscles (Figure 1a); morphological changes in the cervical vertebrae (bony fusion of the posterior elements (Figure 1b)); shortening of the first metacarpal bone (Figure 1c); overall shortening of the second to fifth toes (Figure 1d); and heterotopic ossification of the bilateral shoulder, elbow and hip joints (Figure 1e).

(a): Bilateral heterotopic ossification in the paraspinal muscles (arrow). No scoliosis is observed. (b): Bony fusion of the posterior elements of the cervical vertebrae (left image: X-ray, right image: 3D-CT). (c): Slight shortening of the first metacarpal bones. (d): Overall shortening of toes. No malformation of the great toes was observed. (e): Heterotopic ossification of the right hip joint (arrow), windblown deformity and diffuse osteopenia. 3D-CT, three-dimensional computed tomography.
Written informed consent was obtained from the patient and her family for gene analysis and preparation of this report. The Ethical Committee of The University of Tokyo approved this study. Genetic diagnosis was performed at the Project of Clinical and Basic Research for FOP at Saitama Medical University. All exons of ACVR1 were amplified by a standard PCR method using Pfx platinum DNA polymerase (Invitrogen, Carlsbad, CA, USA). The PCR product that was purified by a Microcon-100 column (Takara Bio Shiga, Japan) was directly sequenced using an ABI3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Results
Analysis was performed to verify the ACVR1 (p.Arg206His) mutation; however, it was not identified. All exons of ACVR1 were then examined, leading to the identification of the ACVR1 (c.774G>T; p.Arg258Ser) mutation in exon 5 of ACVR1 (Figure 2), which is the same mutation reported by Ratbi et al. and Eresen-Yazıcıoğlu et al.

(a) The c.774G>T mutation of ACVR1. Analysis by direct sequence identified the ACVR1 (c.774G>T) heterozygous mutation in exon 5 of ACVR1. (b) Genomic conservation among species.
Discussion
We report the clinical and radiological findings of the patient with FOP due to a recurrent mutations of ACVR1, c774G>T (p.Arg258Ser). Characteristic findings of the classic FOP include great toe malformation from the time of birth and progressive heterotopic ossification of the muscle and the surrounding tissue until ~10 years of age. However, reports of an FOP variant without great toe malformation have recently appeared.6,9,12,16
There have been two previous reports of patients with p.Arg258Ser caused by c.774G>T, but neither had great toe malformation. Ratbi et al.14 reported a patient due to c.774G>T (p.Arg258Ser), whose onset of heterotopic ossification at 8 years of age, and other signs included short first metatarsals and exostosis of different sizes involving the dorsal and lumbar vertebrae, distal segment of the left femur and proximal segment of the left tibia that were observed in the X-rays. Eresen-Yazıcıoğlu et al.15 reported the same mutation, c.774G>T (p.Arg258Ser), in a patient with FOP, whose onset of heterotopic ossification occurred at 10 years of age, and ROM was restricted in the temporomandibular, shoulder, elbow and knee joints. X-rays revealed kyphosis of the thoracic vertebrae and lumbar lordosis, and thinning of the scalp hair was also observed. Both two patients did not have great toe malformation.
Bocciardi et al.9 reported additional two unrelated patients with FOP, who had a same amino-acid alteration (p.Arg258Ser) but a different nucleotide alteration of c.774G>C.9 Phynotypic difference between c.774G>T and c.774G>C is the presence of great toe malformation. Great toe malformation was not observed in any patients with c.774G>T while it was not a common factor in patients with c.774G>C as one of two patients in fact had malformation of the great toes. The patient (FOP12) had no great toe deformity, and onset of ectopic ossification was observed at the age of 4 years because of painful swelling in the vertebral region, and the patient (FOP12) did not experience another flare-up until the age of 18 years. On the other hand, the patient (FOP17) had great toe malformation, and the onset of heterotopic ossification was at 14 years of age.
Clinical manifestations of the patients with FOP due to p.Arg258Ser of ACVR1 were summarized in Table 1 together with those of the patients due to the common ACVR1 mutation (p.Arg206His). The clinical features of the present patient resemble those reported by Ratbi et al. and Eresen-Yazıcıoğlu et al. in that there was no great toe malformation, and the clinical course demonstrated a somewhat delayed onset compared with classic FOP. The difference included no obvious spinal deformity and overall shortening of the toes, and no ossification was observed in the biopsy that was performed at the site of swelling. We believe that the lack of obvious spinal deformity was due to the late onset of heterotopic ossification and because the ossification was relatively symmetrical. The observation that ossification did not occur after the biopsy is an important difference between this variant and classic FOP. However, the progression of decreased activities of daily living after onset was comparatively faster than those in previous reports.
Patients with c.774G>T and c.774G>C lead to the same amino-acid change p.Arg258Ser. It has been known that the mutation which result in same amino-acid change is insignificant to phenotypic differences because such changes in DNA would not alter the composition of the proteins encoded by genes. But recently, there have been reports that in some cases it can still result in altered function because synonymous mutations can alter protein folding.17,18
Meanwhile, comparison of phenotypic difference among five patients is too few to support the hypothesis of the genotype–phenotype correlation. Therefore, possibility of other factors including environmental factors and other genomic modifiers must also be taken into consideration.
In FOP, the clinical symptoms, mutations and mechanism of onset are gradually being discovered. Moreover, it has become evident that the location of mutation differentiates the clinical symptoms from typical to atypical FOP features. This report is extremely significant in terms of providing new evidence of symptoms experienced by patients with c.774G>T who present with clinical findings that are different from those of c.774G>C but in whom mutation occurs in the same amino acid. Accumulating data on novel mutations is important for evaluating pathology, establishing treatments, and contributing to clarify in vivo mechanisms of p.Arg258Ser and its relationship with other mutation types in future studies.
References
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38: 525–527.
Connor JM, Evans DA . Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br 1982; 64: 76–83.
Lin GT, Chang HW, Liu CS, Huang PJ, Wang HC, Cheng YM . De novo 617 G-A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva. J Hum Genet 2006; 51: 1083–1086.
Nakajima M, Haga N, Takikawa K, Manabe N, Nishimura G, Ikegawa S . The ACVR1 617 GNA mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva. J Hum Genet 2007; 52: 473–475.
Lee DY, Cho TJ, Lee HR, Park MS, Yoo WJ, Chung CY et al. ACVR1 gene mutation in sporadic Korean patients with fibrodysplasia ossificans progressiva. J Korean Med Sci 2009; 24: 433–437.
Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 2009; 30: 379–390.
Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG et al. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS ONE 2009; 4: e5005.
Smith R, Russell RG, Woods CG . Myositis ossificans progressiva. Clinical features of eight patients and their response to treatment. J Bone Joint Surg Br 1976; 58: 48–57.
Bocciardi R, Bordo D, Di DM, Di RM, Ravazzolo R . Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet 2009; 17: 311–318.
Carvalho DR, Navarro MM, Martins BJ, Coelho KE, Mello WD, Takata RI et al. Mutational screening of ACVR1 gene in Brazilian fibrodysplasia ossificans progressiva patients. Clin Genet 2010; 77: 171–176.
Furuya H, Ikezoe K, Wang L, Ohyagi Y, Motomura K, Fujii N et al. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H). Am J Med Genet Part A 2008; 146A: 459–463.
Gregson CL, Hollingworth P, Williams M, Petrie KA, Bullock AN, Brown MA et al. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011; 48: 654–658.
Hüning I, Gillessen-Kaesbach G . Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation. Mol Syndromol 2014; 5: 201–211.
Ratbi I, Borcciadi R, Regragui A, Ravazzolo R, Sefiani A . Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva. Clin Rheumatol 2010; 29: 119–121.
Eresen-Yazıcıoğlu C, Karatosun V, Kızıldağ S, Ozsoylu D, Kavukçu S . ACVR1 gene mutations in four Turkish patients diagnosed as fibrodysplasia ossificans progressiva. Gene 2013; 515: 444–446.
Barnett CP, Dugar M, Haan EA . Late-onset variant fibrodysplasia ossificans progressiva leading to misdiagnosis of ankylosing spondylitis. Am J Med Genet Part A 2011; 155: 1492–1495.
Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science 2007; 315: 525–528.
Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B . Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014; 156: 1324–1335.
Acknowledgements
This study was supported in part by the Health and Labour Sciences Research Grants for Research on Measures for Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan, and the Research on Development of New Drugs from Japan Agency for Medical Research and Development, AMED. The genetic diagnosis was performed at the Project of Clinical and Basic Research for FOP at Saitama Medical University (Drs M Matsuoka and K Ikebuchi).
Grant sponsor: Health and Labour Sciences Research Grants for Research on Measures for Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Nakahara, Y., Suzuki, R., Katagiri, T. et al. Phenotypic differences of patients with fibrodysplasia ossificans progressive due to p.Arg258Ser variants of ACVR1. Hum Genome Var 2, 15055 (2015). https://doi.org/10.1038/hgv.2015.55
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.55
