
Abstract
We report a germline nonsense mutation within the extracellular domain of the RING finger ubiquitin ligase RNF43, segregating with a severe form of serrated polyposis within a kindred. The finding provides evidence that inherited RNF43 mutations define a familial cancer syndrome.
Similar content being viewed by others

The serrated polyposis syndrome comprises multiple epithelial polyps in the colon and rectum of serrated histology. WHO clinical criteria1 are the presence of >20 serrated polyps throughout the colon, or >5 proximal to the rectum. Serrated polyps, particularly large sessile polyps in the proximal colon, frequently exhibit the oncogenic V600E mutation together with hypermethylation of the mismatch repair protein MLH1 and are responsible for 15–20% of sporadic colorectal cancer (CRC).2
The serrated polyposis syndrome is associated with CRC risk. Serrated polyposis shows familial clustering,3 and first and second-degree relatives of index cases with serrated polyposis without CRC are more likely to have been diagnosed with CRC or pancreatic cancer.4 The risk of CRC in relatives is higher in those cases diagnosed below the age of 50 years.4 A small number of serrated polyposis patients harbor dominant germline mutations in mismatch repair proteins or biallelic MUTYH mutations, however, when these patients are excluded the familial risk of CRC remains4 and the genetic basis for familial serrated polyposis has not been established. The appearance of serrated polyposis in consanguineous kindreds and in monozygotic twins5 has led to the hypothesis that serrated polyposis may be due in part to recessive or codominant mutations.5
As serrated polyposis is a relatively newly described condition, its natural history is not known and the lifetime risk of CRC in serrated polyposis is also not known. Significantly, it is also not known whether those first and second-degree relatives of serrated polyposis cases, who had a history of CRC or pancreatic cancer, themselves had serrated polyposis.4 If so, then a dominant mode of inheritance in at least a subset of serrated polyposis is likely. If not, however, serrated polyposis could be the result of several codominant alleles.
We identified a severely affected kindred with serrated polyposis. The proband developed microsatellite instability (MSI)– CRC age 23 years arising from a serrated polyp, in the setting of multiple (>50) large serrated polyps throughout the colon; one sibling has multiple serrated polyps and another a single large adenoma. Their mother developed pancreatic cancer and died before this study at age 50 years. On the paternal side, an informative pedigree had no history of multiple polyposis or CRC in the father’s generation. Full sequencing of the MUTYH gene in the proband did not reveal any abnormality.
We performed exon capture and deep sequencing in this kindred to identify strong and potentially interacting cancer alleles.
Exome sequencing
DNA library preparation and exome enrichment was performed at the Australian Phenomics Facility, Australian National University. Input DNA was extracted from saliva, analyzed for integrity then fragmented by mechanical shearing (Covaris AFA). The Agilent XT2 Human, all exon, V5.0 kit and referenced reagents (Agilent, Santa Clara, CA, USA) were utilized for DNA library preparation and exome enrichment. Libraries were indexed and pooled in batches of six before capture. Enriched libraries were sequenced as paired end reads (100 bp runs) on an Illumina HiSeq 2000 at the Australian Cancer Research Foundation (ACRF) Biomolecular Resource Facility at the John Curtin School of Medical Research, Australian National University.
Bioinformatics
Sequence reads were mapped to the GRCh37 assembly of the reference human genome using the default parameters of the Burrows–Wheeler Aligner.6 Untrimmed reads were aligned allowing a maximum of two seed mismatches with repetitively aligned reads discarded. Sequence variants were identified with SAMtools7 and classified as novel, rare (mean allele frequency/MAF⩽0.02), or common (MAF>0.02). Variants were overlapped to ENSEMBL v75 exons and splice site coordinates and annotated using the Ensembl Variant Effect Predictor (VEP)8 to obtain PolyPhen29 and SIFT10 scores for estimating the effect of amino acid substitutions on protein structure and function. Deleterious SNVs in cancer genes were assessed against the COSMIC database and assigned a value. Variant analysis of sequenced pedigrees (pVAAST)11 was performed treating subject 002 as ‘unaffected’ or ‘affected’.
Validation of candidate deleterious mutations
Genotyping for the specific mutations in the probands and relatives were performed by exon-specific PCR and Sanger sequencing.
Subjects gave written informed consent for the study, which received approval from the ACT Health Research Ethics Committee.
Results
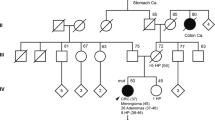
The pedigree of the affected kindred is shown in Figure 1a. The proband II.2, a male nonsmoker of 23, underwent emergency right hemicolectomy for an obstructing CRC and multiple synchronous polyps were palpated at operation. The primary tumor was staged T4N0MX. Immunohistochemistry for the mismatch repair genes was normal. The somatic KRAS codon 12 and 13 mutation panel (COSMIC ID: 516, 517, 518, 520, 521, 522 and 532) was negative. Sequencing of the MUTYH gene in the germline was normal. A female sibling II.3, age 27 years, underwent screening colonoscopy where >20 large serrated polyps were identified. After endoscopic failure to achieve endoscopic clearance of polyps the subject underwent elective subtotal colectomy; >60 polyps were present in the resection specimen. A third sibling II.1, age 21 years, had a single adenoma of the rectum; a second colonoscopy at 1 year was normal.

(a) Pedigree with closed symbols, affected; open symbols, unaffected; gray symbol, deceased. (b) Summary of combined results from whole exome capture and sequencing of genomic DNA from subjects I.2, II.1, II.2 and II.3. Mutations were deemed to be rare (MAF<0.02) or novel in reference to dbSNP. The number of missense, splice site and nonsense single-nucleotide variants (SNV) are shown assuming II.2 and II.3 are affected, and I.2 and II.1 are unaffected. (c) Sanger sequencing of genomic DNA from extant members of the kindred. (d) Conservation of region encompassing arginine converted to stop codon by nonsense mutation. (e) Summary of RNF43 gene structure. Mutated residue is indicated (red triangle) located within the extracellular domain.
We performed whole-exome capture and sequencing (median depth for four exomes was 99×). Variant analysis of sequenced pedigrees was performed treating subject II.1 as ‘unaffected’ or ‘affected’. Rare and novel variant analysis identified 49 variants that conformed to an autosomal dominant inheritance pattern. Of these, 9 were novel variants and 40 were rare (MAF<0.02), and only one was a nonsense variant, which was a novel (R132X) mutation in RNF43 (Figure 1b). This allele was found in subjects II.2 and II.3. Separately, 42 rare or novel variants were identified irrespective of inheritance pattern. Of these, 20 nonsense mutations were identified of which only the previously identified missense RNF43 mutation was segregated with the serrated polyposis phenotype in the pedigree. We confirmed the RNF43 nonsense mutation by Sanger sequencing (Figure 1c).
The mutant allele detected in subjects II.2 and II.3 was present on the minus strand, chromosome 17q22 coordinates 56431037 to 56494931 and represent a C>T transversion at cDNA position 2350 within exon 2 (g.54014C>T). The residue is highly conserved in mammals and birds (Figure 1d), and is located within a well-conserved region that encodes the extracellular protein domain. As a result of this premature stop codon, mutant mRNA is predicted to undergo nonsense mediated decay.
RNF43 encodes a transmembrane RING finger ubiquitin ligase. Amongst its substrates are Wnt agonist frizzled receptors, which are targeted to the lysosome for degradation;12 a mutation in the RING finger domain that inactivated E3 ligase activity increased Wnt signaling.13
Complete Sanger sequencing of RNF43 in each extant member of the pedigree revealed one additional coding variant g.56656C>T in II.3. This missense substitution encodes P231L, and is not predicted to be damaging. Twenty-three instances of homozygosity for this variant have been reported in the TGP database. No somatic mutations replicating this variant have been reported in cancer samples.
We did not determine expression of either the mutant allele or the paternal allele in neoplastic tissue in subjects II.2 or II.3. We expect little or no expression of the mutant allele owing to nonsense mediated decay. As a candidate tumor suppressor gene, RNF43 may be subject to allelic loss or somatic inactivation of the wild-type allele, which is a question for further scrutiny.
RNF43 germline mutations (encoding R113X) were amongst several germ line mutations reported recently in 2 of 20 unrelated sporadic serrated polyposis cases.14 Recently, Giannakis et al.15 described somatic mutations in RNF43 occurring in 18.9% of 185 CRC cases, 17.6% of an independent cohort of 222 CRC cases and 18.1% of 248 endometrial cancer. The majority of RNF43 somatic mutations reported were truncating events (including three instances of R132X), and were strongly associated with MSI-positive cancers and negatively associated with APC mutations, leading Giannakis et al. to propose that mismatch repair deficiency leads to a permissive environment for the acquisition of RNF43 mutations. Taken together with the prior report of Gala et al., our data indicate that RNF43 truncations may instead be pathogenic in the serrated polyposis-cancer sequence, which is independent of the canonical APC mutant adenomatous polyp and in which MSI, when it occurs, arises owing to somatic methylation of the MLH1 gene.16 Furthermore, our report of evidence of heterozygous RNF43 mutations segregating with serrated polyposis within a kindred raises the possibility that this represents a new familial cancer syndrome.
References
References
Lanspa SJ, Ahnen DJ, Lynch HT . Serrated polyposis: the last (or only the latest?) frontier of familial polyposis? Am J Gastroenterol 2012; 107: 779–781.
Jass JR . Serrated adenoma of the colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol 2005; 2: 398–405.
Boparai KS, Reitsma JB, Lemmens V, van Os TA, Mathus-Vliegen EM, Koornstra JJ et al. Increased colorectal cancer risk in first-degree relatives of patients with hyperplastic polyposis syndrome. Gut 2010; 59: 1222–1225.
Win AK, Walters RJ, Buchanan DD, Jenkins MA, Sweet K, Frankel WL et al. Cancer risks for relatives of patients with serrated polyposis. Am J Gastroenterol 2012; 107: 770–778.
Young J, Jenkins M, Parry S, Young B, Nancarrow D, English D et al. Serrated pathway colorectal cancer in the population: genetic consideration. Gut 2007; 56: 1453–1459.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F . Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010; 26: 2069–2070.
Flanagan SE, Patch AM, Ellard S . Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers 2010; 14: 533–537.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D . MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 8: 575–576.
Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, Durtschi JD et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 2014; 32: 663–669.
de Lau W, Peng WC, Gros P, Clevers H . The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev 2014; 28: 305–316.
Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012; 488: 665–669.
Gala MK, Mizukami Y, Le LP, Moriichi K, Austin T, Yamamoto M et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014; 146: 520–529.
Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014; 46: 1264–1266.
Rosty C, Walsh MD, Walters RJ, Clendenning M, Pearson SA, Jenkins MA et al. Multiplicity and molecular heterogeneity of colorectal carcinomas in individuals with serrated polyposis. Am J Surg Pathol 2013; 37: 434–442.
Data Citations
Taupin, Douglas HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.582 (2015)
Acknowledgements
This work was supported by the The Canberra Hospital Private Practice Trust Fund (DT, MC), National Health and Medical Research Council Program Grant 1016953 (CCG and MCC) and NHMRC Australia Fellowship 585490 (CCG, TDA, MF). The authors thank the subjects who gave their time.
Author information
Authors and Affiliations
Contributions
DT and MC and designed the research. DT, MC, BW, YZ, LM, DA, MF and WL performed the research. DR contributed research participants and clinical information. DT, MC, DA, MF and CG were responsible for bioinformatics analysis. DT and MC wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Taupin, D., Lam, W., Rangiah, D. et al. A deleterious RNF43 germline mutation in a severely affected serrated polyposis kindred. Hum Genome Var 2, 15013 (2015). https://doi.org/10.1038/hgv.2015.13
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.13
This article is cited by
-
Inherited BRCA1 and RNF43 pathogenic variants in a familial colorectal cancer type X family
Familial Cancer (2024)
-
Comment on Balsamo et al.: Birt-Hogg-Dubé syndrome with simultaneous hyperplastic polyposis of the gastrointestinal tract: case report and review of the literature
BMC Medical Genomics (2022)
-
Clinical Management of Oligopolyposis of Unknown Etiology
Current Treatment Options in Gastroenterology (2021)
-
Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis
Familial Cancer (2020)
-
Hereditary or Not? Understanding Serrated Polyposis Syndrome
Current Treatment Options in Gastroenterology (2019)
