
Abstract
The role of ecological and changing environmental factors in the radiation of species diversity is a fundamental question in evolutionary biology. Of particular interest is the potential for these factors to determine the boundary between what we would consider differentiation among populations and incipient speciation. Dolphins in the genus Delphinus provide a useful test case, exhibiting morphological variation in beak length, coloration and body size across their wide geographic distribution, and in particular among coastal and more pelagic habitats. Two species have been proposed, D. delphis and D. capensis, but morphologically similar allopatric populations are not monophyletic, indicating that the mostly coastal ‘long-beaked’ D. capensis form is not a single globally distributed species. However, the sympatric populations in the Eastern North Pacific currently designated as these two species are both morphologically and genetically differentiated. Here we use microsatellite DNA and mitochondrial DNA markers to investigate the evolutionary mechanisms that led to this incipient speciation event. We used coalescent and assignment methods to investigate the timing and extent of reproductive isolation. Our data indicate that although there is some level of on-going gene flow, the putative species found in the Eastern North Pacific are reciprocally monophyletic. The timing of isolation appears to be associated with regional changes in paleoceanographic conditions within the Holocene timeframe.
Similar content being viewed by others


Introduction
In the relatively homogeneous marine environment, cryptic diversity among populations or incipient species has been described for a range of taxa from sessile invertebrates (Landry et al., 2003) to fish (Bernardi and Lape, 2005) and marine mammals (Moura et al., 2013). Resource specializations promote intraspecific differential niche use and may lead to genetic structure if gene flow is also restricted, for example, through assortative mating, physical separation within local environments or by divergent selection (Rundle and Nosil, 2005; Nosil, 2012). In marine fishes, both niche partitioning and mate choice were affected by divergent selection on characters that resulted in sympatric speciation within the genus Hexagrammos (Crow et al., 2010). Among cetacean species, both killer whales (Orcinus orca) and bottlenose dolphins (Tursiops truncatus) show genetic differentiation among populations correlated to foraging specializations or habitat dependence (O. orca, Hoelzel et al., 2007; Moura et al., 2014; T. truncatus, Hoelzel et al., 1998; Natoli et al., 2005; Segura et al., 2006; Charlton-Robb et al., 2011; Moura et al., 2013).
A fairly consistent theme among delphinid species is phenotypic differentiation between coastal and pelagic forms that is commonly associated with body size and skull morphology. There is also often genetic differentiation between coastal and pelagic populations, such as for the bottlenose dolphins in the North Atlantic and North Pacific (T. truncatus; see, for example, Natoli et al., 2005; Segura et al., 2006; Lowther-Thieleking et al., 2015) and for the eastern tropical Pacific pantropical spotted dolphins (Stenella attenuata; Escorza-Treviño et al., 2005). In some cases ecological phenotypes associated with habitat have been assigned specific status, such as the pelagic (T. truncatus) and coastal (Tursiops aduncus) forms of bottlenose dolphins in Asia (Wang et al., 1999) and Australasia (Möller and Beheregaray, 2001), and the riverine (Sotalia fluvitalis) and coastal (Sotalia guianensis) forms of the ‘tucuxi’ dolphin (Caballero et al., 2007).
For the genus Delphinus, morphological variation across their wide geographic distribution has complicated the assignment of alpha-taxonomy. More than 20 nominal species of the genus Delphinus had been historically described (Hershkovitz, 1966). Although only D. delphis was formally recognized until the mid 1990s (Evans, 1994), the coastal long-beaked form off California had been proposed to be the species D. bairdii by Dall (1873). The existence of a long-beaked form of common dolphin in this region was later supported by Banks and Brownell (1969). However, the occurrence and description of long-beaked common dolphins elsewhere (for example, D. capensis, Gray, 1828) led to the synonymy of D. bairdii and D. capensis (Van Bree and Purves, 1972). Later, morphological and, for populations in the Eastern North Pacific (ENP), genetic differences reinforced the recognition of D. capensis as a distinct ‘long-beaked’ species in the genus Delphinus (Heyning and Perrin, 1994; Rosel et al., 1994).
Within the ENP, the two named species, D. delphis (hereafter short-beaked ENP (sbENP)) and D. capensis (hereafter long-beaked ENP (lbENP)), can be distinguished based on morphological as well as genetic characteristics (see below). Elsewhere in the world there are long-beaked populations (of the ‘capensis’ type) proposed to occur off Peru, Chile, Venezuela, Brazil, Argentina, South Africa, Madagascar, India, Indonesia, China, Korea and southern Japan (see Hammond et al., 2008; Jefferson et al., 2008), although for a number of these locations occurrence is not well documented. Based on the ‘rostral index’ (rostrum length/zygomatic width ratio, see below), Cunha et al. (2015) identified 5 ‘delphis’ and 9 ‘capensis’ types off Brazil and Argentina among the 14 skulls they measured, and Tavares et al. (2010) identified 47 of 59 measured skulls from this region as the ‘capensis’ type. However, Cunha et al. (2015) suggest that D. capensis should not be used to refer to the long-beaked form, and instead the lbENP form should again be recognized as D. bairdii and all other populations recognized as D. delphis.
Analyses of mitochondrial DNA (mtDNA) and nuclear amplified fragment length polymorphism markers supported the distinction between morphotypes in the ENP (Rosel et al., 1994; Kingston and Rosel, 2004; Kingston et al., 2009). However, independent studies comparing populations elsewhere could not find support for two genetic lineages representing distinguishable ‘long-beaked’ and ‘short-beaked’ species worldwide based on 199 samples analyzed for microsatellite DNA and mtDNA control region sequences (Natoli et al., 2006), mtDNA cytochrome b (cytb) sequences for 343 samples and nuclear sequence data at 5 loci for 90 samples (Amaral et al., 2012) and mtDNA cytb sequences for an additional 37 samples (Cunha et al., 2015). Instead, it was suggested that the long- and short-beaked phenotypes showed convergent adaptation to coastal and pelagic habitat, respectively (Natoli et al., 2006), divergent selection with incomplete lineage sorting (Amaral et al., 2012) or phenotypic plasticity within the species D. delphis (Cunha et al., 2015). A third morphotype with an even longer rostrum and greater number of teeth was identified in the Indian Ocean and off China, and was nominally identified as a subspecies of D. capensis, D. capensis tropicalis (Jefferson and van Waerebeek, 2002). This putative subspecies is genetically differentiated from the two proposed species (Amaral et al., 2012).
In the ENP, the short-beaked common dolphin sbENP is the most common pelagic odontocete (estimated abundance: 2 34 430–4 89 826 in the California current system; Barlow and Forney, 2007) whereas long-beaked common dolphins (lbENP) are more common within the Gulf of California and less abundant overall (4833–43 765; Gerrodette and Palacios, 1996; Carretta et al., 2011). It is in the ENP among the populations of these two forms where genetic differentiation is most pronounced (Rosel et al., 1994; Natoli et al., 2006; Amaral et al., 2012). We focused on this region using robust sample sets and both nuclear and mtDNA markers to better understand the mechanisms that may be supporting incipient speciation and to assess the timing and pattern of differentiation within this genus. We test the hypothesis that local changes in habitat availability during the Holocene coincide with the timing of divergence and consider the implications for the worldwide biogeography of this and related species.
Materials and methods
Sample collection and analysis
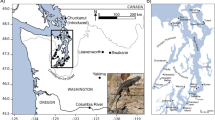
A total of 310 samples were collected from the Gulf of California and off the western coast of Baja California and California (Figure 1; hereafter referred to as the ENP). Dolphins could be assigned to type in the field based on total length (the sbENP form is shorter) and a conspicuously longer beak in the lbENP. Furthermore, the unique crisscross color pattern formed by interaction of the dorsal overlay and cape creates a four-stripe coloration that is distinct in the two forms. In the sbENP form the thoracic patch is more yellowish, the subcape stripe tends to be narrow and faint at the anterior and the flipper stripe tends to narrow and pass low below the corner of the gape (Perrin, 2009). In the lbENP form the pattern is more muted, the spinal field may be grayish, the thoracic patch tends to be pale buff, the flipper-to-annus stripe tends to be broad anteriorly and may be pronounced and contiguous with the flipper stripe and the flipper stripe tends to wander toward the corner of the gape before passing ventrally to join the lip patch mark (Perrin, 2009). For samples from skeletal materials, the ratio of rostral length to zygomatic width is greater in lbENP and can be used as a diagnostic feature (Heyning and Perrin, 1994).

Geographic location of individual samples used in this study. Long-beaked form (lbENP): yellow (light gray) circles, short-beaked form (sbENP; D. delphis): blue (dark gray) circles. Circles often represent more than one dolphin individual sampled in the same location. A full color version of this figure is available at the Heredity journal online.
Skin samples were obtained using standard projectile biopsy sampling (Kellar et al., 2013) from animals found stranded (N=11) or bycaught in fisheries (N=8). An additional 19 tooth samples were obtained from museum specimens (see Supplementary Table S1). DNA was extracted from biopsies following standard phenol chloroform or salt precipitation protocols (Sambrook and Russell, 2001). DNA from tooth samples was extracted using spin columns (Qiagen, Manchester, UK) in an ancient DNA facility to prevent cross-contamination, and included both PCR and extraction controls. Fragments of 778 bp from the mtDNA control region were amplified from 49 D. delphis and 132 lbENP samples (sample subsets chosen at random), and 480 bp was sequenced from 11 of the tooth samples (the full length could be sequenced from 8 of them). Sixteen microsatellite DNA loci were amplified from 173 D. delphis and 137 lbENP samples. See Supplementary Table S2 for methodological details. Sex was determined by amplifying fragments of the genes Zfy/x and SRY (Pomp et al., 1995).
Data analysis
Mitochondrial DNA sequences were aligned using CLUSTAL X (Thompson et al., 1994) and haplotypes identified using DNAsp version 3 (Rozas and Rozas, 1999). The best evolutionary model for the mtDNA data was assessed using MODELTEST 3.7 (Posada and Crandall, 1998). Inbreeding coefficients (FST and φST), haplotype diversity (h), nucleotide diversity (π), Nei’s pairwise genetic distance, Tajima’s D and Fu’s Fs neutrality tests were estimated using ARLEQUIN 2.0 (Schneider et al., 2000). Comparisons against database mtDNA sequences were for 277 bp overlapping sequence (within the hypervariable region 1 (HVR1)) for samples from the Pacific Ocean (Rosel et al., 1994), South Australia and Tasmania (Bilgmann et al., 2008), North Atlantic, South Africa and Argentina (Natoli et al., 2006), applying the Kimura 2-parameter model for φST analysis (closest option available in ARLEQUIN to the transition model suggested in MODELTEST).
Phylogenetic relationships among mtDNA haplotypes were examined with a median-joining network (MJN) with T. truncatus as an outgroup, generated in the program NETWORK 4.5.1.0 (Bandelt et al., 1999). The MJN was calculated using only haplotype sequences of the same length (778 bp) to prevent artificial clustering. Historical demographic expansion was investigated using mismatch distributions (Rogers and Harpending, 1992). An index of time since expansion expressed in mutational time (τ=2μt, where μ is the mutation rate/generation and t is time in generations), was estimated by a generalized nonlinear least square approach using ARLEQUIN 2.0 (Schneider et al., 2000).
Genotypes for 16 microsatellite loci were tested for the presence of allelic dropout and null alleles using MICRO-CHECKER (Van Oosterhout et al., 2004) and checked to confirm that all samples represented unique individuals. Observed and expected heterozygosity, Nei’s genetic distance, FST and deviation from Hardy–Weinberg equilibrium were assessed in ARLEQUIN 2.0 (Schneider et al., 2000). Allelic richness and FIS were assessed using FSTAT 2.9.3 (Goudet, 2002). Tests for sex-biased dispersal (based on sex-biased differences in assignment probabilities and measures of diversity) were implemented in FSTAT 2.9.3. Clustering assignments were performed in five independent runs for each prior estimate of population number (K= 1–5) as implemented in STRUCTURE 2.3.4 (Pritchard et al., 2000) using the correlated allele frequency and admixture models, with 106 repetitions after a burn-in of 105 (chosen after initial trials to confirm stabilization of parameters). Possible migration ancestry was tested using the admixture model with prior population (morphotype) information assuming a migration rate of v=0.01.
Isolation, migration and historical demography
Recent migration rates were estimated using BayesAss (Wilson and Rannala, 2003). The Markov chain Monte Carlo was run for 9 00 000 iterations of burn-in and of 3 000 000 iterations for the chain and mixing parameter for migration rates were tested as 0.01 and 0.005, as suggested by Wilson and Rannala (2003). A coalescent approach as implemented in IMa (Hey and Nielsen, 2007) was performed to estimate the time since population divergence, asymmetric migration rates between putative species and effective population sizes (Ne). These parameters were scaled by mutation rate across loci (μ=5 × 10−7 s s y−1 for mtDNA, after Ho et al., 2007; and 5 × 10−4 l y−1 for microsatellite DNA loci, after Whittaker et al., 2003). At least three replicate runs with random seeds were carried out with a burn-in of 105 steps, and 200 chains (M-mode). This approach was run independently for the mtDNA sequence data and with the combination of both mtDNA data and 16 microsatellite loci so that we could consider the matriline data separately. After runs converged (and showed consistently high effective sample size values), we applied the nested model approach in the L-mode of IMa. This model tests parameters estimated in the M-mode based on the likelihood of the nested models (Hey and Nielsen, 2007).
Results
Genetic differentiation
Among the 192 samples analyzed for mtDNA, we identified 106 control region haplotypes, 95 haplotypes of 778 bp and 11 haplotypes of 480 bp (Supplementary Table S3). There is one fixed difference between lbENP and sbENP (D. delphis) populations, a G/A transition in the nucleotide position 213 (Supplementary Table S3). The best fit model of molecular evolution (for the 778 bp sequence incorporating both the HVR1 and the conserved central region) was Tamura–Nei (Tamura and Nei, 1993) with a γ-distribution of 0.073. Haplotype and nucleotide diversities were marginally lower in lbENP (H=0.961±0.0069, π=0.0088±0.0046, N=131) compared with sbENP (H=0.993± 0.0058, π=0.0142±0.0073, N=49). No shared haplotypes were observed between them, and genetic differentiation was highly significant (FST=0.024, P<0.00001; φST=0.526, P<0.00001). The comparison among published data from nine other populations around the world (including both long-beaked and short-beaked populations) showed no consistent differentiation pattern among populations, as indicated in an earlier study based on fewer populations and much smaller sample sizes from the ENP (Natoli et al., 2006; Figure 2 and Supplementary Table S4). For φST the lbENP population in our study stands out as the most differentiated by a large margin (Figure 2), and equally differentiated from long and short-beaked populations elsewhere. The lbENP population is also most distinct for measures of pairwise difference and Nei’s genetic distance (Supplementary Figure S2). The South Africa population stands out for FST (which considers only haplotype frequencies; Supplementary Table S4), and the lowest haplotype diversity is also found there (haplotypes/sample=0.40 compared with 0.42–0.93 elsewhere).

Heat map comparison of φST values based on 277 bp mtDNA control region comparing Galicia, Spain (Gal), the Celtic Sea, Ireland (Cel), Scotland (Sco), Madeira (Mad), the western North Atlantic (WNA), South Africa (lbSA), Argentina (lbArg), Tasmania (Tas), the short-beak population in southern and Baja California (sbENP) and the long-beak population in southern and Baja California (lbENP). Putative long-beaked population names are in blue (gray) text. A full color version of this figure is available at the Heredity journal online.
Allelic richness at microsatellite DNA loci was higher in sbENP than lbENP (mean=13.09, s.d.=4.56 and mean=10.63, s.d.=3.61, respectively; see Supplementary Table S5). For all loci combined, there were 65 private alleles for sbENP and 12 for lbENP. There were null alleles detected for EV14 and EV37 and significant departures from Hardy–Weinberg equilibrium at EV14, EV37, KWM2a, KWM2b and TexVet5 (though actual differences between Ho and He were small, see Supplementary Table S5). Analyses omitting these 5 loci showed equivalent results (data not shown), and hence results for 16 loci are reported. Genetic differentiation between lbENP and sbENP was low but significant (FST=0.029, P<0.001 based on 16 microsatellite loci), consistent with mtDNA findings. STRUCTURE also strongly supported differentiation, and the highest hierarchical level of structure (ΔK) was K=2 (Figure 3 and Supplementary Figure S3). The lnP(K) value for K=3 was marginally higher and showed some evidence for substructure within the sbENP population (Supplementary Figure S4), but without any clear geographic correspondence (and therefore not clearly supported). This test also showed evidence of two potential introgression events (indicated with an asterisk, Figure 2). These individuals were identified as F1 migrants, given the proportional contribution (P) of D. delphis to their nuclear genotypes (Hap 93, P=0.505 and Hap 89, P=0.877). Both putative hybrids were females with mtDNA haplotypes diagnostic for lbENP and the phenotypic characteristics of the long-beaked form (both skull samples, based on rostral length and zygomatic width ratio after Heyning and Perrin, 1994), but with nuclear DNA assignments to sbENP (D. delphis). These events suggest the introgression of D. delphis males with lbENP females. Our analyses in FSTAT testing for sex-biased dispersal were not significant (data not shown), although these tests have relatively low power.

Structure assignments where K=2 and based on an a priori migration rate estimate of 0.01 (* represents potential events of introgression).
Population demographic parameters and phylogenetic analyses
Contemporary gene flow estimated using BayesAss was asymmetric, indicating more gene flow from sbENP into lbENP populations (lbENP→sbENP: m=0.006 proportional representation, confidence interval: 0.003–0.017; sbENP→lbENP: m=0.023, confidence interval: 0.009–0.042). Estimates of gene flow using IMa (either based on mtDNA or both mtDNA and microsatellite DNA) showed a similar magnitude and directionality (see Table 1 for details). The nested analysis in L-mode (based on both marker types) showed that the best supported models included migration (unidirectional into lbENP in the maximum likelihood model), and only the single least supported model did not (Table 2). IMa estimates of contemporary female effective population sizes (based on mtDNA; Table 1a) indicated that the sbENP population size is considerably larger than lbENP. The small ancestral population size is unexpected (as ancestral Ne is often inflated in the two population–one ancestor model because of current or ancestral connectivity with other populations). The estimates for the time of divergence between sbENP and lbENP based on mtDNA only suggested that the diversification began at the start of the Holocene ~10 000 years ago (Table 1a). When microsatellite DNA data were included, estimates for splitting time became more recent, and migration rate estimates somewhat higher (Table 1b).
Mismatch distributions based on 778 bp mtDNA sequences were not significantly different from the expansion models (Supplementary Figure S1; sbENP: P=0.33; lbENP: P=0.30). Harpending’s raggedness index (Rogers and Harpending, 1992) was small and nonsignificant (sbENP: H=0.003, P=0.77; lbENP: H=0.011, P=0.69). For sbENP, tau was 10.58 (95% confidence interval: 7.57–12.28) and the estimated time of expansion was 13 599 YBP (years before present) (9730–15 784). For lbENP, tau was 11.05 (3.11–15.28) and the expansion time 14 203 (3997–19 640). The MJN phylogenetic reconstruction shows distinct clusters for lbENP and sbENP (D. delphis) haplotypes, with evident differences in the structure of the two clusters (Figure 4). There is complex reticulation at the center of the sbENP network, suggesting unsampled haplotypes, whereas lbENP showed a number of star-shaped structures, typical of an expansion signal. For Tajima’s D, neither sbENP (D=−1.29, P=0.07) nor lbENP (D−0.80, P=0.21) were significant, but Fu’s Fs was highly significant for both populations (sbENP: Fs=−24.51 P<0.00001; lbENP: Fs=−24.84, P<0.00001).

MJN of the 95 mtDNA control region haplotypes (778 bp). Circle diameter is proportional to haplotype frequency. Yellow (light gray) circles correspond to lbENP and blue (dark gray) circles to sbENP (D. delphis). T. truncatus (TT, in red/gray) was used as the outgroup. A full color version of this figure is available at the Heredity journal online.
Discussion
The process of speciation is rarely observed and poorly understood. However, local adaptation is likely an important mechanism (see Lenormand, 2012). Increasingly, the molecular mechanisms supporting local adaptations are being identified (see Savolainen et al., 2013), and evolutionary change can be over very short timeframes (for example, from industrial melanism in Biston betularia or the influence of an el Niño cycle on Geospiza fortis; Lamichhaney et al., 2015; Van’t Hof et al., 2016). Phenotypic differentiation among populations and species inhabiting coastal and offshore aquatic environments is well established, and sometimes clearly associated with adaptive differentiation (see Schluter, 2000). For the genus Delphinus, there are coastal populations worldwide that show morphological differentiation associated with longer beak length among other characteristics, although there are also some short-beaked populations found nearshore (for example, in the Mediterranean Sea), and stable isotope analysis of beach-cast specimens was used to question the distribution of long-beaked forms off North Africa (Pinella et al., 2011). However, as we confirm here, only the long-beaked coastal population off southern California and Baja California is strongly genetically differentiated (Figure 2 and Supplementary Table S4). Across the broader geographic range there may be convergent evolution of morphotype for the long-beaked form, but not at the same stage of genetic differentiation (c.f., Heyning and Perrin, 1994; Rosel et al., 1994; Natoli et al., 2006). Cunha et al. (2015) have suggested that beak length could be phenotypically plastic, although this will be difficult to test for a dolphin species. As has been often proposed, it is possible that both plasticity and local adaptation play a role (see West-Eberhard, 2003). These issues address a key question in evolutionary biology: under what conditions does intraspecific differentiation lead to incipient speciation?
In our study there is clear evidence for significant genetic differentiation between the parapatric (and partially sympatric; Figure 1) sbENP and lbENP populations off southern California and Mexico, consistent with the earlier assessment by Rosel et al. (1994). However, there is also evidence for continuing gene flow. Two putative hybrids were detected and both IMa and BayesAss suggested biased gene flow from the sbENP to the lbENP form at non-zero levels. This is not necessarily inconsistent with incipient speciation, as hybrids between recognized species are common for cetaceans (see Crossman et al., 2016), and incipient speciation is possible even in the face of recurrent or continuous gene flow (Hey, 2006; Niemiller et al., 2008), for example, among three divergent forms of Tennessee cave salamander, Gyrinophilus palleucus (Niemiller et al., 2008). Incomplete lineage sorting could explain some missassigned individuals based on a given marker, but the congruence of phenotype and different marker types for the same individuals makes this interpretation less likely. The evidence for very recent gene flow (identified F1 and current migrants) suggests a recent separation, as do the divergence time estimates.
The MJN reconstruction showed two well-defined lineages, but lineage structure differed for lbENP compared with the sbENP form. For sbENP there was extensive reticulation consistent with a large, poorly sampled population with a broad distribution. This, together with the high estimate of female Ne for this population and relatively high diversity (consistent with relative diversity levels reported by Natoli et al., 2006 and Amaral et al., 2012 worldwide) suggests that D. delphis (sbENP in our study region) represents the parent population from which lbENP was founded. The lbENP lineage in contrast shows a series of star formations and a relatively small number of common haplotypes. This suggests that the population is well represented by the sample set, and that it has undergone a recent expansion. The mismatch distributions were also consistent with a recent (since the last glacial maximum) expansion for both populations. Some IMa runs suggested the opposite effect, but the two population, one ancestor model can overestimate ancestral Ne (see Hey and Nielsen, 2004). Overall, the network structure together with directional gene flow from D. delphis to lbENP suggests the isolation of a divergent coastal population. The mismatch distributions suggest that this happened sometime after the last glacial period (given the applied mutation rate).
The IMa analyses estimated a time of divergence (based on mtDNA and a mutation rate of 5 × 10−7 s s y−1) of 12 033 YBP for female lineages. Mutation rates vary among species (see, for example, Lambert et al., 2002; de Bruyn et al., 2009; Phillips et al., 2009), and there is no precise estimate for this species. However, we used a rate that falls within the intervals of those derived from time series data and a coalescent approach for other vertebrate species (see Phillips et al., 2009). For example, in the Adélie penguin, Pysocelis adeliae, 9.6 × 10−7 s s y−1 (Lambert et al., 2002), southern elephant seal, Mirounga leonina, 9.8 × 10−7 s s y−1 (de Bruyn et al., 2009) and Steller sea lion, Eumetopias jubatus, 2.7 × 10−7 s s y−1 (Phillips et al., 2009). These estimates are based on HVR1 and our control region sequence is somewhat longer, but we get similar estimates when just the HVR1 region is used (data not shown). If the mutation rate was an order of magnitude slower, the time of divergence would be an order of magnitude older. A previous study used a rate of 9.86 × 10−9 for the cytb mtDNA locus to conclude that divergence between sbENP and lbENP took place during the Pleistocene (Amaral et al., 2012). In a comparison between control region and cytb mutation rates in cetaceans, Alter and Palumbi (2009) suggested that the control region evolved 4–5 times faster than cytb. Using the time series calibrations we applied would suggest a rate of ∼1 × 10−7 for cytb, roughly an order of magnitude faster than the rate used in the earlier study. This difference explains our different conclusions about divergence time.
When microsatellite DNA markers were included together with mtDNA, a splitting time of 167–2621 YBP was estimated and migration rate estimates became higher. Sun et al. (2012) show that for humans, microsatellite DNA mutations tend toward the center of the size distribution, and that this bias may affect divergence time estimates by up to a factor of two compared with a strict stepwise mutation model (as applied here for the microsatellite DNA loci), and hence the true splitting time values could be higher. Another possible factor would be ongoing male-mediated gene flow and the cessation of female dispersal, as suggested by our data, that would reduce the apparent division time when nuclear markers were included. Male-biased dispersal is common in mammals (see, for example, Dobson, 1982), although in this case the implication is that only males disperse.
Therefore, our data credibly suggest a division date after the last glacial maximum. The Gulf of California and western margin of California and Baja California have experienced active geological and paleoclimatic changes proposed to have driven the evolution of several taxa (Jacobs et al., 2004). During the Pleistocene–Holocene transition, ~15 000–10 000 YBP, warm anomalies of sea surface temperatures occurred leading to the collapse of the California Current (Herbert et al., 2001). The typical conditions of the California Current, as currently persist, were reestablished during the Holocene ~9000 YBP, resulting in higher regional productivity (Ortiz et al., 2004). As a consequence of rising sea level, a favorable emerging coastal habitat became available, creating the environment for a founder dolphin population to exploit this new resource and become independent of the offshore source population. To the extent that Holocene changes in current patterns also had an effect on the pelagic environment, high productivity and therefore food availability could have promoted a demographic expansion in the source D. delphis population as well, as suggested by the data.
We propose that a group founded from the ancestral D. delphis population took advantage of emerging habitat, reducing resource competition and leading to resource specialization and a consequent reduction in gene flow with the parental population. A similar scenario has been proposed for South Australian and South-eastern Tasmanian common dolphins where Pleistocene changes in the ecosystem and the emergence of the Bassian land-bridge may have promoted genetic isolation and differentiation (Bilgmann et al., 2008), but in that case there is no evidence for incipient speciation. There are examples of marine mammal species responding quickly to environmental change to exploit emerging habitat (see, for example, de Bruyn et al., 2009), but a key question is why this local Delphinus population in the ENP shows greater progression toward speciation than other parapatric and sympatric populations of common dolphin morphotypes elsewhere in the world? The process seems to have advanced further for Tursiops populations and sibling species in similar habitats (see Moura et al., 2013).
In the broader context, we propose that the timing of events would be consistent with a response to changing habitat and local adaptation to differential resources. There are now some well-established examples of ecological differentiation/speciation (see review in Yoder et al., 2010), although relatively few studies have explored the role of habitat release following climate change. One example involved the differential evolution of grasswrens in the Amytornis textilis modestus complex after the appearance of novel shrubland habitat during the late Plio-Pleistocene (Norman and Christidis, 2016). Soria-Carrasco et al. (2014) showed that stick insects (Timena cristinae) adapting to novel host plant species showed parallel changes at the genomic level, revealing the central role of natural selection in that system, although it is clear that plasticity is sometimes important as well, likely in combination with selection (such as among feeding morphs of some salmonid freshwater fish in littoral compared with benthic habitat; see Klemetsen, 2010).
The data presented here show for the first time how local processes associated with adaptation to emerging habitat associated with the paleoceanography of the California Current could have led to incipient speciation in this genus, and suggest that other regional populations distinguished by morphotype may be independently undergoing similar processes. Other Delphinus populations may be at a different stage in the process, in an environment where isolation is less strongly promoted, or even represent the collapse of a previous pattern of differentiation (for example, as seen for whitefish, Coregonus lavaretus; Bhat et al., 2014). Chance events associated with founder size and the extent of genetic drift, or differences associated with environmental conditions or local processes, may be relevant to the differential extent of isolation in different regions. However, although we cannot fully resolve those mechanisms, our data provide a clear signal that these are likely local independent processes that started in the relatively recent past (suggested by our data to be in association with the transition from the last glacial period), as opposed to a more ancient process that predated the global distribution of differentiated sibling species. We propose an ecological process of differentiation and, in the ENP, incipient speciation that is driven by environmental factors associated with climate change.
Data accessibility
DNA sequences: Genbank accessions HE680096–HE680202; microsatellite DNA genotypes: Dryad: doi:10.5061/dryad.q1t87.
References
Alter SE, Palumbi SR . (2009). Comparing evolutionary patterns and variability in the mitochondrial control region and cytochrome b in the three species of baleen whales. J Mol Evol 68: 97–111.
Amaral AR, Beheregaray LB, Bilgmann K, Freitas L, Robertson K, Sequeira M et al. (2012). Influences of past climatic changes on historical population structure and demography of cosmopolitan marine predator, the common dolphin (genus Delphinus. Mol Ecol 21: 4854–4871.
Bandelt HJ, Forster P, Röhl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Banks RC, Brownell RL . (1969). Taxonomy of the common dolphins of the Eastern Pacific Ocean. J Mamm 50: 262–271.
Barlow J, Forney KA . (2007). Abundance and population density of cetaceans in the California Current ecosystem. Fish Bull 105: 509–526.
Bernardi G, Lape J . (2005). Tempo and mode of speciation in the Baja California disjunct fish species Anisotremis davisonni. Mol Ecol 14: 4085–4096.
Bhat S, Amundsen PA, Knudsen R, Gjelland KØ, Fevolden SE, Bernatchez L et al. (2014). Speciation reversal in European whitefish (Coregonus lavaretus (L.)) caused by competitor invasion. PLoS One 9: e91208.
Bilgmann K, Moller LM, Harcourt RG, Gales R, Beheregaray LB . (2008). Common dolphins subject to fisheries impacts in Southern Australia are genetically differentiated: implications for conservation. Anim Cons 11: 518–528.
Caballero S, Trujillo F, Vianna JA, Barrios-Garrido H, Montiel MG, Beltran-Pedreros S et al. (2007). Taxonomic status of the genus Sotalia: species level ranking for "tucuxi" (Sotalia fluviatilis and "costero" (Sotalia guianensis dolphins. Mar Mammal Sci 23: 358–386.
Carretta JV, Chivers SJ, Perryman WL . (2011). Abundance of the long-beaked common dolphin (Delphinus capensis in California and western Baja California waters estimated from a 2009 ship-based line transect survey. Bull S Calif Acad Sci 3: 152–164.
Charlton-Robb K, Gershwin L, Thompson R, Austin J, Owen K, McKechnie S . (2011). A new dolphin species, the Burrunan Dolphin Tursiops australis sp. nov., endemic to Southern Australian coastal waters. PLoS One 6: e24047.
Crossman CA, Taylor EB, Barret-Lennard LG . (2016). Hybridization in the Cetacea: widespread occurrence and associated morphological, behavioral and ecological factors. Ecol Evol 6: 1293–1303.
Crow KD, Munehara H, Bernardi G . (2010). Sympatric speciation in a genus of marine reef fishes. Mol Ecol 19: 2089–2105.
Cunha HA, Lozizaga de Castro R, Secchi ER, Crespo EA, Lailson-Brito J, Azavedo AF et al. (2015). Molecular and morphological differentiation of common dolphins (Delphinus sp.) in the Southwestern Atlantic: testing the two species hypothesis in sympatry. PLoS One 10: e0140251.
Dall WH . (1873) Field Notes. Smithsonian Transcription Service. Available at: http://edan.si.edu/transcription/pdf_files/6973.pdf.
de Bruyn M, Hall BL, Chauke LF, Baroni C, Koch PL, Hoelzel AR . (2009). Rapid response of a marine mammal species to holocene climate and habitat change. PLoS Genet 5: e1000554.
Dobson FS . (1982). Competition for mates and predominant juvenile male dispersal in mammals. Anim Behav 30: 1183–1192.
Escorza-Treviño S, Archer EI, Rosales M, Lang AR, Dizon AE . (2005). Genetic differentiation and intraspecific structure of Eastern Tropical Pacific spotted dolphins, Stenella attenuata, revealed by DNA analyses. Cons Gen 6: 587–600.
Evans WE . (1994) Common dolphin, white bellied porpoise Delphinus delphis Linnaeus. In Ridgway SH, Harrisons R (eds) Handbook of Marine Mammals. Academic Press Ltd: London UK/San Diego, pp 191–224.
Gerrodette T, Palacios DM . (1996) Estimates of cetacean abundance in EEZ waters of the eastern tropical Pacific SWFSC Admin. Rep. No LJ-96-10: 28pp.
Goudet J . (2002). FSTAT 2.9.3.2. Institute of Ecology, Switzerland.
Gray JE . (1828) Original figures and short systematic descriptions of new and unfigured animals. Spicilegia Zoologica Part 1 1–2.
Hammond PS, Bearzi G, Bjørge A, Forney K, Karczmarski L, Kasuya T et al. (2008). Delphinus capensis. The IUCN Red List of Threatened Species 2008: e.T6337A12663800.
Herbert TD, Schuffert JD, Andreasen D, Heusser L, Lyle M, Mix A et al. (2001). Collapse of the California Current during glacial maxima linked to climate change on land. Science 293: 71–76.
Hershkovitz P . (1966). Catalog of living whales. Bull US Nat Mus 246: 1–259.
Hey J . (2006). Recent advances in assessing gene flow between diverging populations and species. Curr Opin Genet Dev 16: 592–596.
Hey J, Nielsen R . (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760.
Hey J, Nielsen R . (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA 104: 2785–2790.
Heyning J, Perrin W . (1994). Two forms of common dolphins (genus Delphinus from the eastern North Pacific; evidence for two species. Contr Sci 1–35.
Ho SYW, Kolokotronis SO, Allaby RG . (2007). Elevated substitution rates estimated from ancient DNA sequences. Biol Lett 3: 702–705.
Hoelzel AR, Hey J, Dahlheim ME, Nicholson C, Burkanov V, Black N . (2007). Evolution of population structure in a highly social top predator, the killer whale. Mol Biol Evol 24: 1407–1415.
Hoelzel AR, Potter CW, Best PB . (1998). Genetic differentiation between parapatric “nearshore” and “offshore” population of the bottlenose dolphin. Proc R Soc B 265: 1177–1183.
Jacobs DK, Haney TA, Louie KD . (2004). Genes, diversity, and geologic process on the Pacific coast. Ann Rev Earth Plan Sci 32: 601–652.
Jefferson TA, van Waerebeek K . (2002). The taxonomic status of the nominal dolphin species Delphinus tropicalis van Bree, 1971. Mar Mammal Sci 18: 787–818.
Jefferson TA, Webber MA, Pitman RL . (2008) Marine Mammals of the World; A Comprehenisize Guide to their Identification. Academic Press: New York, pp 592.
Kellar NM, Trego ML, Chivers SJ, Archer FI, Minich JJ, Perryman WL . (2013). Are there biases in biopsy sampling? Potential drivers of sex ratio in projectile biopsy samples from two small delphinids. Mar Mamm Sci 29: E366–E389.
Kingston SE, Adams LD, Rosel PE . (2009). Testing mitochondrial sequences and anonymous nuclear markers for phylogeny reconstruction in a rapidly radiating group: molecular systematics of the Delphininae (Cetacea: Odontoceti: Delphinidae). BMC Evol Biol 9: 245.
Kingston SE, Rosel PE . (2004). Genetic differentiation among recently diverged delphinid taxa determined using AFLP markers. J Hered 95: 1–10.
Klemetsen A . (2010). The charr problem revisited: exceptional phenotypic plasticity promotes ecological speciation in postglacial lakes. Freshw Rev 3: 49–74.
Lambert DM, Ritchie PA, Millar CD, Holland B, Drummond AJ, Baroni C . (2002). Rates of evolution in ancient DNA from Adelie penguins. Science 295: 2270–2273.
Lamichhaney S, Berlund J, Almen MS, Maqbool K, Grabherr M, Martinez-Barrio A et al. (2015). Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518: 371–375.
Landry C, Geyer LB, Arakaki Y, Uehara T, Palumbi SR . (2003). Recent speciation in the Indo-West Pacific: rapid evolution of gamete recognition and sperm morphology in cryptic species of sea urchin. Proc R Soc B 270: 1839–1847.
Lenormand T . (2012). From local adaptation to speciation: specialization and reinforcement. Int J Ecol 2012: 508458.
Lowther-Thieleking JL, Archer FI, Lang AR, Weller DW . (2015). Genetic differentiation among coastal and offshore common bottlenose dolphins, Tursiops truncatus, in the eastern North Pacific Ocean. Mar Mamm Sci 31: 1–20.
Moura AE, Kenny JG, Chaudhuri R, Hughes MA, Welch A, Reisinger RR et al. (2014). Population genomics of the killer whale indicates ecotype evolution in sympatry involving both selection and drift. Mol Ecol 23: 5179–5192.
Moura AE, Nielsen SCA, Vilstrup JT, Moreno-Mayar JV, Gilbert MTP, Gray H et al. (2013). Recent diversification of a marine genus (Tursiops spp. tracks habitat preference and environmental change. Syst Biol 62: 865–877.
Möller LM, Beheregaray LB . (2001). Coastal bottlenose dolphins from southeastern Australia are Tursiops aduncus according to sequences of the mitochondrial DNA control region. Mar Mamm Sci 17: 249–263.
Natoli A, Birkun A, Aguilar A, Lopez A, Hoelzel AR . (2005). Habitat structure and the dispersal of male and female bottlenose dolphin (Tursiops truncatus. Proc R Soc B 272: 1217–1226.
Natoli A, Cañadas A, Peddemors VM, Aguilar A, Vaquero C, Fernández-Piqueras P et al. (2006). Phylogeography and alpha taxonomy of the common dolphin (Delphinus sp.). J Evol Biol 19: 943–954.
Niemiller ML, Fitzpatrick BM, Miller BT . (2008). Recent divergence with gene flow in Tennessee cave salamanders (Plethodontidae: Gyrinophilus inferred from gene genealogies. Mol Ecol 17: 2258–2275.
Norman JA, Christidis L . (2016). Ecological opportunity and the evolution of habitat preferences in an arid-zone bird: implications for speciation in a climate-modified landscape. Sci Rep 6: 19613.
Nosil P . (2012) Ecological Speciation. Oxford Univ Press.
Ortiz JD, O'Connell SB, DelViscio J, Dean W, Carriquiry JD, Marchitto T et al. (2004). Enhanced marine productivity off western North America during warm climate intervals of the past 52 ky. Geology 32: 521–524.
Perrin WF . (2009) Common dolphins. In: Perrin WF, Wursig B, Thewissen JGM (eds) Encyclopedia of Marine Mammals. Elsevier: USA, pp 249–255.
Phillips CD, Trujillo RG, Gelatt TS, Smolen MJ, Matson CW, Honeycutt RL et al. (2009). Assessing substitution patterns, rates and homoplasy at HVRI of Steller sea lions, Eumetopias jubatus. Mol Ecol 18: 3379–3393.
Pinella AM, Borrell A, Aguilar A . (2011). Common dolphin morphotypes: niche segregation or taxonomy? J Zool 284: 239–247.
Pomp D, Good BA, Geisert RD, Corbin CJ, Conley AJ . (1995). Sex identification in mammals with polymerase chain reaction and its use to examine sex effects on diameter of day-10 or 11 pig embryos. J Anim Sci 73: 1408–1415.
Posada D, Crandall KA . (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rogers A, Harpending H . (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9: 552–569.
Rosel PE, Dizon AE, Heying JE . (1994). Genetic analysis of sympatric morphotypes of common dolphins (genus Delphinus. Mar Biol 59–167.
Rozas J, Rozas R . (1999). DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175.
Rundle H, Nosil P . (2005). Ecological speciation. Ecol Lett 8: 336–352.
Sambrook J, Russell DW . (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press: New York.
Savolainen O, Lascoux M, Merila J . (2013). Ecological genomics of local adaptation. Nat Rev Genet 14: 807–819.
Schluter D . (2000) The Ecology of Adaptive Radiation. Oxford University Press.
Schneider S, Roessli D, Excoffier L . (2000). ARLEQUIN ver. 2.0. A Software for Population Genetics Data Analysis. U. G. Genetics and Biometry Lab, editor. Available at: http://cmpg.unibe.ch/software/arlequin3/.
Segura I, Rocha-Olivares A, Flores-Ramirez S, Rojas-Bracho L . (2006). Conservation implications of the genetic distinction of Tursiops truncatus ecotypes in the Gulf of California. Biol Cons 133: 336–346.
Soria-Carrasco V, Gompert Z, Comeault AA, Farkas TE, Parchman TL, Johnston JS et al. (2014). Stick insect genomes reveal natural selection’s role in parallel speciation. Science 344: 738–742.
Sun JX, Helgason A, Masson G, Ebenesersdottir SS, Li H, Mallick S et al. (2012). A direct characterisation of human mutation based on microsatellites. Nat Genet 44: 1161–1165.
Tamura K, Nei M . (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526.
Tavares M, Moreno B, Siciliano S, Rodriguez D, Santos M, Lailson-Brito J et al. (2010). Biogeography of common dolphins (genus Delphinus in the Southwestern Atlantic Ocean. Mamm Rev 40: 40–64.
Thompson JD, Higgins DG, Gibson TJ . (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680.
Van Bree PJH, Purves PE . (1972). Remarks on the variability of Delphinus bardii (Cetacea: Delphinidae). J Mamm 53: 372–374.
Van Oosterhout C, Hutchinson WF, Willis DPM, Shipe P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 536–538.
Van’t Hof AE, Campagne P, Rigden DJ, Yung CJ, Lingley J, Quail MA et al. (2016). The industrial melanism mutation in British peppered moths is a transposable element. Nature 534: 102–105.
Wang JY, Chou LS, White BN . (1999). Mitochondrial DNA analysis of sympatric morphotypes of bottlenose dolphins (genus: Tursiops in Chinese waters. Mol Ecol 8: 1603–1612.
West-Eberhard MJ . (2003) Developmental Plasticity and Evolution. Oxford University Press.
Whittaker JC, Harbord RM, Boxall N, Mackay I, Dawson G, Sibly RM . (2003). Likelihood-based estimation of microsatellite mutation rates. Genetics 164: 781–787.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Yoder JB, Clancey E, Des Roches S, Eastman JM, Gentry L, Godsoe W et al. (2010). Ecological opportunity and the origin of adaptive radiations. J Evol Biol 23: 1581–1596.
Acknowledgements
We thank Dr Axayacátl Rocha-Olivares, Dra Gisela Heckel, MenC Jorge Montano and Oc Alejandra Baez from the Centro de Investigación Científica y de Educación Superior de Ensenada (CICESE) for the logistic support to conduct fieldwork; Kelly Robertson, Aimee Mann and Juan Carlos Salinas from the Southwest Fisheries Science Center-NOAA, the SWFSC tissue archive and their fishery observer program, and Dra Diane Gendron from the Centro de Interdisciplinario de Ciencias Marinas (CICIMAR) for sample contribution and helpful feedback. We also thank the Colección Nacional de Mamíferos of Instituto de Biología-UNAM, the Laboratorio de Mamíferos Marinos-Facultad de Ciencias-UNAM and Instituto Tecnológico y de Estudios Superiores de Monterrey for allowing access to collection specimens. We thank Andre Moura from Durham University for developing multiplex PCR profiles. We also thank the CONACYT abroad studies scholarship program awarded to Iris Segura-García, Rudolf-Small Grants for research, Durham University-Ustinov College and School of Biological and Biomedical Sciences for funding.
Author contributions
IS-G carried out the fieldwork, molecular lab work and data analyses; ARH and IS-G designed the project and wrote the manuscript; ARH contributed to data analysis; JPG collected samples and helped draft the manuscript; SC and RD-G helped with data acquisition. All authors gave final approval for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Segura-García, I., Gallo, J., Chivers, S. et al. Post-glacial habitat release and incipient speciation in the genus Delphinus. Heredity 117, 400–407 (2016). https://doi.org/10.1038/hdy.2016.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.66
This article is cited by
-
Iterative Habitat Transitions are Associated with Morphological Convergence of the Backbone in Delphinoids
Journal of Mammalian Evolution (2022)
-
Eco-Evolutionary Processes Generating Diversity Among Bottlenose Dolphin, Tursiops truncatus, Populations off Baja California, Mexico
Evolutionary Biology (2018)
