
Abstract
The mountains of southwest China (MSC) harbor extremely high species diversity; however, the mechanism behind this diversity is unknown. We investigated to what degree the topography and climate change shaped the genetic diversity and diversification in these mountains, and we also sought to identify the locations of microrefugia areas in these mountains. For these purposes, we sampled extensively to estimate the intraspecific phylogenetic pattern of the Chinese mole shrew (Anourosorex squamipes) in southwest China throughout its range of distribution. Two mitochondrial genes, namely, cytochrome b (CYT B) and NADH dehydrogenase subunit 2 (ND2), from 383 archived specimens from 43 localities were determined for phylogeographic and demographic analyses. We used the continuous-diffusion phylogeographic model, extensive Bayesian skyline plot species distribution modeling (SDM) and approximate Bayesian computation (ABC) to explore the changes in population size and distribution through time of the species. Two phylogenetic clades were identified, and significantly higher genetic diversity was preserved in the southern subregion of the mountains. The results of the SDM, continuous-diffusion phylogeographic model, extensive Bayesian skyline plot and ABC analyses were congruent and supported that the Last Interglacial Maximum (LIG) was an unfavorable period for the mole shrews because of a high degree of seasonality; A. squamipes survived in isolated interglacial refugia mainly located in the southern subregion during the LIG and rapidly expanded during the last glacial period. These results furnished the first evidence for major Pleistocene interglacial refugia and a latitudinal effect in southwest China, and the results shedding light on the higher level of species richness in the southern subregion.
Similar content being viewed by others


Introduction
During climate change, a species will shift its distribution to remain in a suitable habitat, or adapt to a new environmental conditions (Hewitt, 2004). The latter is not common, as most species tend to exhibit niche conservatism (Wiens and Graham, 2005). The shifts in distribution are usually accompanied by changes in population size and genetic diversity. For this reason, the Pleistocene climate fluctuations had strong effects on the genetic diversity and geographical structure of many species by repeated shifting of its distribution (Hewitt, 1996). This effect could be more complicated in an area with complex topography, such as the mountains of southwest China (hereafter referred to as MSC).
The MSC, also known as the Hengduan Mountains, and the adjacent areas have complex topographies that support extremely high biodiversity, and have been recognized as biodiversity hotspots (He and Jiang, 2014). One plausible explanation for the great biodiversity is known as the ‘refugia hypothesis’ (Zhang, 2002). The concept of refugia used here is ‘climate change refugia’, including glacial refugia for temperate-adapted taxa and interglacial refugia for cold-adapted taxa (Stewart et al., 2010). Refugia can also be classified as macrorefugia or microrefugia (i.e., cryptic refugia), depending on the size (Bennett and Provan, 2008). In the MSC, the complex topography provides ecologically diverse habitats, so that organisms could find suitable habitats by moving up and down the mountains during periods of unfavorable climate conditions, with lower extinction risks. An alternative hypothesis suggested that the complex topography created fragmented habitats in the mountains, so that populations of a species were isolated, resulting in allopatric diversification (He and Jiang, 2014).
Recent intraspecific phylogeographic studies of endemic species have revealed pre-Pleistocene diversifications, spatial genetic structures and demographic changes (He and Jiang, 2014). These results supported that large river valleys acted as geographic barriers for small terrestrial animals, and there were multiple refugia in the mountains where populations survived during the Pleistocene glaciations. However, the drainage system appeared to have had variable effects (from promotion to depression) on different species. The effect of climate change varied with latitude and topography, and each species responded individually because of differences in their dispersal ability and habitat affinities (He and Jiang, 2014). Thus, testing the genetic structure of other endemic species is still necessary. Moreover, the locations of macro- and microrefugia are still unknown. This problem could be resolved by using extensive sampling to examine the genetic pattern of a widely distributed species. In the present study, we selected the Chinese mole shrew (Anourosorex squamipes Milne-Edwards, 1872), a small insectivoran mammal as our focal species (Supplementary Figure S1).
The living Asian mole shrews include four species in the genus Anourosorex, which are small and semifossorial mammals. A. squamipes is mainly distributed in southwestern China, inhabiting diverse habitats including forest, farm land and wasteland. This species occupies a wide range of elevations from 300 to 4000 m a.s.l. and latitudes from 18°N to 35°N. The genus has rich fossil records in China and Japan from the late Miocene to the Pleistocene strata, which is represented by eight species (Supplementary Figure S2; Storch et al., 1998). In contrast, a molecular study suggested that A. squamipes has an evolutionary history of only 0.66 million years (myr) (1.35–0.18; He et al., 2010). The contradicting shallow evolutionary time and ancient fossils indicated either a massive extinction or undiscovered cryptic lineages. The living mole shrews are relic species of the tribe Anourosoricini, which were widely distributed in Eurasia during the Miocene (Wojcik and Wolsan, 1998). Climate changes have played an important role in the evolution of this tribe according to at least three lines of evidence: (1) fossils from the late Miocene and early Pliocene suggested that anourosoricines were not tolerant to low levels of precipitation or soil humidity (van Dam, 2004); (2) the global cooling and desiccating near the Pliocene/Pleistocene boundary resulted in the extinction of the tribe in Europe and only Anourosorex survived in East and Southeast Asia (Storch et al., 1998); and (3) a molecular phylogeographic study based on a mitochondrial gene suggested that the genetic structure of Anourosorex yamashinai in Taiwan was shaped by climate change-driven, range expansions/contractions and consequent secondary contacts, supporting an interglacial scenario (Yuan et al., 2006). Therefore, it would be interesting to explore why these animals survived in MSC and examine how climate change affected the evolution of A. squamipes.
Over the past 20 years, 383 specimens of A. squamipes were collected throughout its distribution range in southwest China and adjacent areas. By including available sequences from GenBank, 389 complete cytochrome b (CYT B) and 311 NADH dehydrogenase subunit 2 (ND2) sequences were included to investigate genetic diversity and phylogeographic patterns. Species distribution modeling (SDM), continuous-diffusion phylogeographic and statistical coalescent analyses were also used. Our objects were to determine: (1) whether cryptic lineages existed in MSC; (2) whether the mountain and drainage system shaped the genetic structure; (3) whether Pleistocene climate changes affected genetic diversity and diversification patterns; (4) whether MSC refugia existed during the climatic fluctuations, and if so, where the macro- and microrefugia were located.
Materials and methods
Ethics statement
The sample collection did not require any official permit from the Chinese government, as these animals are considered as pests. Animal scarifying was approved by the Ethics Committee of Kunming Institute of Zoology, Chinese Academy of Sciences, China.
Sampling and sequencing
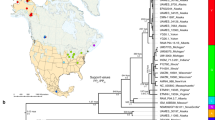
A total of 383 samples of A. squamipes were collected from 44 localities in the Qinling Mountains, Hengduan Mountains, Sichuan Basin and Yunnan Plateau (Figure 1b and Supplementary Table S1). Genomic DNA was extracted from either liver or muscle samples using the DNeasy Tissue Kit (Qiagen, Hilden, Germany) or the phenol/proteinase K/sodium dodecyl sulfate method. Two complete mitochondrial loci (CYT B and ND2) were amplified. The primers are given in Supplementary Table S2. All PCR products were purified and sequenced in both directions on an ABI 3730 Genetic Analyzer Beijing (Tiangen, Beijing, China).

Phylogenetic tree of A. squamipes (a) and sample localities of A. squamipes (b). The Bayesian tree and the ML tree topologies are very similar, thus only the Bayesian tree is represented. Branch numbers represent PPs/bootstrap values. In the distribution map, numbers are corresponding to those in the Supplementary Table S1 and presented as pie charts. Slice size is proportional to the frequency of the subclades occurring in the site.
The sequenced amplicons were assembled and edited using DNASTAR Lasergene v.7.1. Additional CYT B and ND2 sequences of A. squamipes and A. yamashinai obtained in the previous studies were also included (Ohdachi et al., 2006; He et al., 2010). Each locus was aligned using MUSCLE (Edgar, 2014). Premature stop codons were examined by eye. We first estimated the CYT B and ND2 gene trees independently and cross-checked the phylogenetic positions of each specimens on both trees (data not shown) to ensure there was no cross-specimen contamination.
Phylogenetic analyses and molecular dating
We estimated maximum-likelihood (ML) trees using RAxML v.7.4.4 (Stamatakis et al., 2008) and Bayesian trees using BEAST v.1.7.5 (Drummond et al., 2012). The best-fit partitioning scheme and evolutionary model for each partition were determined in PartitionFinder v.1.0.0 (Lanfear et al., 2012) under the Bayesian information criterion. We defined the data blocks based on the genes and codon positions, and PartitionFinder determined a six partition scheme (i.e., each codon position of each gene should be given a specific model). The RAxML did not allow any models other than GTR+CAT and GTR+Γ (Stamatakis et al., 2008), thus we selected GTR+Γ for all partitions. We used the rapid bootstrapping algorithm and ran 500 bootstrap replicates. The ML tree reconstructions were performed on the CIPRES Science Gateway.
For BEAST analyses, the evolutionary models of the first, second and third codon positions of CYT B were K80+G, TrN+G and HKY+G, respectively. The evolutionary models of the first, second and third codon positions of ND2 were TrN+G, HKY+G and HKY+G, respectively. Each BEAST analysis used a random starting tree, birth–death tree prior, an uncorrelated log-normal relaxed molecular clock for each codon position (Ho and Lanfear, 2010) and the program’s default prior distributions of model parameters. Analyses were run for 50 million generations, and were sampled every 5000 generations. The analyses were repeated two times and convergence was assessed using TRACER v.1.5. For divergence time estimation, a reduced data set of 134 CYT B haplotypes was used. We calibrated the most recent common ancestor (MRCA) of A. squamipes and A. yamashinai in consideration of the phylogenetic relationships, fossil records and the divergence time from a previous study (He et al., 2010). Rich fossils representing six Anourosorex species were found in southern China (Storch et al., 1998). The oldest fossil of A. squamipes was from strata of the middle Pleistocene in Anhui Province, a location out of its current distribution range (0.3 Ma; Huang et al., 1995). Further, analyses of two nuclear genes (ApoB and RAG1) revealed that the MRCA of A. squamipes+A. yamashinai was also in the Pleistocene at ~0.66 Ma (1.35–0.18) (He et al., 2010). Because the fossil records of Anourosorex are relatively continuous and the divergence time coincides with the fossil records, we assumed the divergence between A. squamipes and A. yamashinai was at 0.76–0.30 Ma. We applied a log-normal distribution as the prior model for the calibration and enforced the median divergence time to equal 0.66 (mean=0.37, s.d.=0.17, offset=0.3).
Yuan et al. (2006) used an evolutionary rate of 2% per million years, which led to a 3-myr divergence between A. squamipes and A. yamashinai. We also compared our results with this scenario using Bayes factor (BF). We generated a new data set of two nuclear genes (ApoB and BRCA1, downloaded from GenBank) representing 25 soricid species, including A. squamipes and A. yamashinai. The divergence between Soricinae and Crocidurinae at ~20 Ma (Ziegler, 1989) was constrained and used as a ‘checkpoint’ (95% confidence interval (95% CI)=25–20). First, we used 0.66 Ma as the MRCA of A. squamipes+A. yamashinai. Then, we used 3 Ma (s.d.=0.1) as the MRCA of A. squamipes+A. yamashinai. The two analyses were run using BEAST v.1.7.5, and the BF was calculated in Tracer v.1.5. The 3-myr divergence scenario was strongly rejected (2 ln BF>100).
Genetic diversity and geographic pattern
Haplotype and nucleotide (Pi) diversities were calculated using DnaSP v.5 (Librado and Rozas, 2009). The significant levels were estimated by coalescent simulations with 1000 replicates. The population subdivision was estimated using the hierarchical analysis of molecular variance (AMOVA) with Arlequin v.3.5. AMOVA was performed with 10 000 permutations with two grouping options based on the geographical distributions (see Results).
Species distribution modeling
We obtained 209 geographic coordinates. These records were collected in our field work using a Global Positioning System. We subsampled coordinates using the R package Raster v.2.1 to reduce the effect of sampling bias (in our case, more sample localities in the southern subregion). Only one record was retained per grid cell (0.5 × 0.5°).
Climate data, including 19 climate variables for the current condition (~1950–2000; 30 arcsec resolution), Last Interglacial Maximum (~116 000–130 000 years BP; 30 arcsec) and Last Glacial Maximum (LGM) (~21 000 years BP; 2.5 arcmin) were downloaded from the WorldClim website (http://www.worldclim.org/). The climate in the LGM was predicted by the Paleoclimate Modeling Intercomparison Project Phase II and the climate in the LIG was predicted by Otto-Bliesner et al. (2006). The environmental layers were clipped to a region from 70°E to 150°E and from 10°S to 55°N using ArcGis v.10. We used MaxEnt v.3.3.3k for SDM with two sampling strategies (Phillips and Dudik, 2008). First, we used 75% of our coordinates as training data and 25% as test data. We also ran the modeling with 53-fold cross-validations. This approach removed the spatial sorting bias, and should outperform a single training/test split strategy (Phillips and Dudik, 2008). The area under the receiver operating characteristic curve was used to evaluate the accuracy of our models. A good model should have an area under the receiver operating characteristic curve value ranging from 0.9 to 1.0 (Baldwin, 2009). After modeling, we used the MaxEnt's explain tool to investigate how the MaxEnt prediction was determined by the climatic variables in the study area.
Demographic expansions and geographic expansions
The historical population dynamics were analyzed by Fu’s test and Ramos-Onsins and Rozas’s R2 test, mismatch distribution analyses and extended Bayesian skyline plots (Heled and Drummond, 2008). The Fu’s and R2 tests were estimated using DnaSP v.5. Mismatch distribution analyses were performed using Arlequin v.3.5 (Excoffier and Lischer, 2010), and the statistical significance was assessed by 1000 permutations. Tau (τ) was estimated simultaneously with mismatch distribution analyses. If a sudden expansion model was suggested, the expansion time was calculated with the equation τ=2ut (Rogers and Harpending, 1992). We used 1 year as the generation time for A. squamipes.
Extended Bayesian skyline plots were estimated using BEAST v.1.7.5. The median divergence times and the 95% CI from the molecular dating results were used for the calibrations. CYT B and ND2 were both used, and each gene was given an independent log-normal relaxed clock model. The analyses were performed for each subclade (see Results) and repeated at least two times. We used uniform distributions for the mean clock rates (0–1.0) and for the population prior mean (0–100). The analyses were run for 50 million generations and sampled every 5000th generation.
We ran continuous-diffusion phylogeographic analyses using BEAST v.1.7.5. The analyses were performed for clades N and S independently with the CYT B data set. A time homogeneous SRW (Standard Random Walk) model and two time-heterogeneous RRW (Relaxed Random Walk) models (cauchy RRW and gamma RRW) (Lemey et al., 2010) were used. We compared SRW and RRW models using BF estimated with the path and stepping-stone sampling methods (Baele et al., 2012). To calculate path and stepping-stone sampling values, the marginal likelihood of each analysis was estimated with 500 000 steps, with 5000 burn-in. The ancestral locations through time were visualized using Google Earth v.7.0 and SPREAD v.1.0.7rc (Bielejec et al., 2011).
Multispecies coalescent model and approximate Bayesian computation
We used coalescent-based approaches to estimate the evolutionary history by comparing alternative topologies and demographic scenarios. First, we used Bayesian Phylogenetics & Phylogeography (BPP) v.3.1 to estimate the population size parameters for both modern and ancestral species (θs) (Yang and Rannala, 2010). We considered the ND2 and CYT B as a single locus, and performed analyses for clades N (80 sequences) and S (225 sequences) separately, because the software did not accept more than 240 sequences. We used species delimitation=0, species tree=0 as prior, thus the software calculated the posterior distribution of species divergence time parameters (τs) and population size parameters (θs) under the multispecies coalescent model. The topology was constrained based on the results of the BEAST analyses. Gamma prior G (2, 1,000) was used on the population size parameters (θs), and the age of the root in the species tree (τ0) was assigned gamma prior G (2, 2,000). Each rjMCMC was run for 100 000 generations and sampled every 10 generations after discarding 10 000 generations as pre-burn-in. The analyses for each data set were repeated three times. The θs values obtained in the BPP analyses were used as a guidance in the approximate Bayesian computation (ABC) analyses to avoid comparison of an inordinately large number of scenarios.
We used DIYABC v.2.0.4 for the ABC-based scenario comparisons (Cornuet et al., 2014). First, we compared five topologies to test if our data set supported a simultaneous divergence scenario (Supplementary Figures S3a–e). Then, we compared demographic scenarios to test the occurrence of bottlenecks when populations split from their ancestor, following the strategy of Louis et al. (2014). Ten alternative scenarios were compared (Supplementary Figures S3f and o) based on the results of BPP, which found large θs for clades N2 and S5 (see Result). The priors for divergence times, populations sizes and mutations rates are given in Supplementary Table S3. The summary statistics within populations included the number of segregating sites, the mean pairwise differences and its variance, and Tajima’s D. Statistics computed between groups were the mean of within- sample pairwise differences, mean of between sample pairwise differences and Fst between two samples. We simulated 106 data for each run. After each simulation, we first run pre-evaluation to make sure that the scenarios and priors produced simulated data sets that were close enough to the observed data set. Then, we computed the posterior probabilities (PPs) of each scenario and compared scenarios by logistic regression. The logistic regression was performed on linear discriminant analysis components (Cornuet et al., 2014).
Results
Phylogenetic relationships and molecular divergence time
A total of 389 complete CYT B (1140 bp) and 313 complete ND2 (1044 bp) sequences of A. squamipes were used. The new sequences were submitted to GenBank under accession numbers KT032255–KT032946. The phylogeny of Anourosorex was overall finely resolved with most of the relationships being strongly supported (i.e., bootstrap values ⩾70 and PPs ⩾0.95). The Bayesian and ML analyses produced very similar topologies, and no incongruence was observed (Figure 1a). Three major clades were recovered (clades N, S and T); clade N was mainly distributed in the relatively northern area (Sichuan, Gansu and Shaanxi); clade S was distributed in the southern area (Yunnan); clade T was represented by two sequences from Thailand (Figure 1b). Two subclades nested in clades N and five subclades nested S. The BEAST divergence time estimation found clades T and S diverged at ~0.43 Ma (0.53–0.30). The MRCAs of clades N and S were at 0.35 Ma (0.51–0.22) and 0.31 Ma (0.43–0.21), respectively (Figure 2).

Divergence time was estimated using BEAST and a relaxed molecular clock (a), and China Loess Plateau grain size and magnetic susceptibility variations through time (b) (Sun et al., 2006). (a) Branch lengths represent time. Node numbers represent median ages. Node bars indicate the 95% CI for the node age. (b) The grain size and magnetic susceptibility values were standardized. High values correspond to interglacial periods and low values correspond to glacial periods.
Genetic diversity and structure
Hd and Pi diversities are given in Table 1. Although the diversifications in both clades N (mainly distributed in Gansu, Shaanxi and Sichuan; 29°N–34°N) and S (mainly distributed in Yunnan; 20°N–29°N) started at about the same time (0.35–0.31 Ma; Figure 2), the southern clade had higher Hd and Pi. To test whether the higher genetic diversity was because of a larger population size, we subsampled 123 CYT B and ND2 sequences from clade S 1000 times and estimated the Hi and Pi for each subsample data set using the package DnaSP and APE v.3.0 for the R environment (Paradis et al., 2004). One-sample t-tests were used and statistically confirmed that clade S had higher Hd and Pi (P<0.001).
AMOVA analyses were implemented using two grouping options. We first grouped populations into three groups corresponding to the three major clades (N, S and T). Second, we considered rivers as boundaries and grouped populations into nine groups. The results of AMOVA revealed significant genetic structures at all hierarchical levels (P<0.001), and the largest proportion of variances were both found among both groups (Table 2). However, the second grouping option reduced the proportion of variances among groups (65.61% vs 73.62%).
Species distribution modeling
The two sampling strategies produced similar results. In the single training/test split run, the area under the receiver operating characteristic curve values of our model were 0.984 (based on the training data) and 0.986 (based on the test data). For the replicate runs, the mean area under the receiver operating characteristic curve value of our model was 0.980 (s.d.=0.027), suggesting that the potential distribution fits well with our data. Although we only used coordinates of A. squamipes for niche modeling, the predicted geographic ranges also covered the distributions of the other three living species, supporting a niche conservatism scenario (Figure 3a). The precipitation of warmest quarter had the highest percentage contribution to the model (bio18; 23.1%), and the second important variable was the mean temperature of driest quarter (bio9; 18.3%). The temperature seasonality (bio4) had the highest permutation importance (48.2%), which also was supported by the jackknife test. The annual precipitation (bio12) did not strongly contribute to the model (6.0% percent contribution and 0.8% permutation importance). According to our modeling, the suitable habitats in the LGM were similar to the present day but expanded in southwest China and Southeast Asia (Figure 3b). In contrast, suitable habitat in Sichuan, eastern Yunnan and Southeast Asia was heavily reduced in the LIG (Figure 3c). The mountain areas in northwestern Yunnan were the most stable habitats through the LIG to the present day. By using the MaxEnt's explain tool, we found that the expansion in the LGM was mostly attributed to more stable temperature (bio4 (temperature seasonality)), and reduced distribution in the LIG was because of highly variable temperature (bio4) and/or precipitation (bio15 (precipitation seasonality)) (Supplementary Figure S4).

Ecological niche model projections of (a) the current distributions of A. squamipes, (b) the distribution during the LGM and (c) the distribution during the Last Interglacial Maximum (LIG). Sample locations are shown. Locations in red color were used for SDM and locations in violet were not used. A full color version of this figure is available at the Heredity journal online.
Demographic expansion and spatial expansion tests
The Fu’s and R2 tests detected population expansions in clades N and S; however, it was only statistically supported for clade S (P<0.001). The population stability for most subclades, with the exception of subclade S5, could not be statistically rejected (Table 1). Subclade N1 had a significant but not small R2 value. These results coincided with the results of Extended Bayesian skyline plot, which also supported an expansion scenario of subclade S5 (Supplementary Figure S5). According to the equation τ=2ut, the expansions occurred at about 35–13 thousand years ago (Ka) (95% CI=52–4; Table 2). The Extended Bayesian skyline plot analyses suggested that a continuous growth started at about 80 ka. All analyses supported that the demographic expansions occurred after the LIG.
Path and stepping-stone sampling found the Cauchy RRW model had the highest log marginal likelihood values for clades N and S. The resulting BFs were 17–29 for clade N and 22–143 for clade S, strongly supporting variable dispersal rates over time (Supplementary Table S4). The ancestral distribution reconstructions suggested that the MRCAs of clades N and S were in the western Sichuan Mountains and the southern Hengduan Mountains, respectively (Figure 4).

The reconstructed spatiotemporal diffusion of A. squamipes, shown at 230 ka (a), 120 ka (b), 21 ka (c) and the present day (d). Lines show spatial projections of representative phylogenies of the clades N and S. Colored clouds represent statistical uncertainty in the estimated locations (95% CI). A full color version of this figure is available at the Heredity journal online.
Multispecies coalescent model and ABC analyses
The BPP analyses found that N2 had a larger population size compared with N1 and the ancestor of clade N. In the clade S, S5 had the largest population size followed by the ancestor of clade S (Supplementary Table S5). We used a two-step procedure to identify the best evolutionary scenario with DIYABC. In the first step, a simultaneous divergence scenario in both clades N and S had the highest support with a PP=0.96 (95% CI=0.94–0.97; Figure 5a). We further compared demographic scenarios based on the simultaneous divergence topology and the results of BPP. We tested whether bottlenecks occurred at sublineages and their ancestors. The scenarios assuming bottlenecks in the subclades N2 and S5, as well as in the ancestor of clade N (PP=0.25, 95% CI=0.16–0.35; Figure 5b) or bottlenecks in the subclades N2 and S5 (PP=0.23, 95% CI=0.14–0.32; Figure 5c) received the highest supports, followed by the scenario assuming bottlenecks in all subclades and ancestor of clade N (PP=0.14, 95% CI=0.08–0.19).

Schematic diagram of the best evolutionary scenario supported in the first step of DIYABC analyses (a), and the best (b) and the second best (c) scenarios supported in the second step of DIYABC analyses. A complete list of scenarios is given in Supplementary Figure S3.
Discussion
Congruence among different analyses and robustness of SDM
In this study, we integrated genetic and geospatial approaches with extensive sampling through the distribution area of A. squamipes. The results of different analyses were largely congruent, and supported each other. The SDM suggested that the southern subregion had more stable habitats. This result was supported by higher genetic diversity and more lineages in the south. The SDM revealed range shifts through time, and found that the LIG as an unfavorable period. The demographic analyses including extended Bayesian skyline plot- and ABC-based approaches also supported a bottleneck in the LIG and a glacial expansion scenario. Continuous-diffusion phylogeographic analyses also supported dispersal rate changes through time.
For SDM, we only used the coordinates of A. squamipes for modeling, but MaxEnt also successfully predicted the geographic range of the other living species in eastern Himalaya and Taiwan. This result supported a niche conservatism scenario for the living shrew moles and facilitated the interpretation of our results, which shed light on the degree to which the climate and topography shaped genetic diversity and structure.
Shallow phylogeny and spatial genetic structure
Despite the fact that Anourosorex mole shrews had long evolutionary histories of at least 6 myr in southwest China and were represented by rich fossil records, the living species in this area had a relatively shallow phylogeny. Although we used extensive sampling, no cryptic lineage or diversification was detected, which suggests that all fossil species were eliminated during the marked climate change from the late Miocene through the early Pleistocene. The AMOVA analyses suggested that rivers did not act as strong geographic barriers for dispersal. This pattern was different from other terrestrial small mammals (Liu et al, 2012), but similar to a semiaquatic mammal (Yuan et al, 2013). Although very little is known about the behavioral ecology of any mole shrew, the results indicated that A. squamipes was a good swimmer and was able to cross the large rivers.
A. squamipes ecology and response to climate change
van Dam (2004) hypothesized a mean annual precipitation of 600 mm year−1 was the lower limit for anourosoricines. However, this variable (bio4) contributed very little to our modeling. We explored bio4 in LIG, LGM and the present day, and found that in south China, the three periods had annual precipitations higher than 600 mm year−1, which explained why Anourosorex survived in this area.
The SDM found that the pattern of distributional changes in the LIG and LGM analogized an interglacial refugia scenario. An interglacial refugia scenario was also revealed for A. yamashinai from Taiwan (Yuan et al., 2006) and a montane bird (Parus monticolus) from MSC (Wang et al, 2013), both of which inhabit much higher elevations (>1000 m above sea level). However, the current distribution of A. squamipes is more similar to the LGM rather than the LIG, despite the fact that the present day is another (Holocene) interglacial period. By using the MaxEnt's explain tool, we found that the species is sensitive to seasonal climate stability and is not tolerant to severe temperature (bio4) or precipitation (bio15) changes through a year. Although the LGM was colder than the present day, the climate (as predicted by Paleoclimate Modeling Intercomparison Project Phase II) in southwest China was more stable comapred with that in the LIG.
It is not easy to interpret why is A. squamipes not tolerant to seasonal climatic changes, especially because the species inhabits a wide range of elevations (300–4000 m) and habitats. The highest percentage contribution of bio9 and bio18 implied that the species was not sensitive to temperature or precipitation alone, but a combination of the two variables. Moreover, adaptation to seasonal climatic changes requires complex behavioral, anatomica, and physiological adjustments for small mammals (Merritt, 1986). Thus, we hypothesize the mole shrews are able to find suitable microhabitat at any elevations, but have problems finding such an environment during extreme temperature and precipitation changes in a short period, because of their poor dispersal ability. There is very little ecology data available for mole shrews (or any other shrews in China), thus physiological experiments are warranted to test the hypothesis.
Interestingly, P. monticolus also had a similar distributional pattern in the LGM and the present day (Wang et al, 2013), indicating that seasonality may have also played an important role for other species. This finding also implied that increasing seasonality and extreme weather events in the present day may have especially strong influences on species.
Massive extinction and latitude effect
What was responsible for the extinction of fossil mole shrew species and the different genetic diversity in the northern and southern subregions? Our results suggested that the potential distribution have greatly shifted during climate change. In addition, the Pliocene and Pleistocene encompassed climatic cycles of differing amplitudes and durations (see Figure 2b), the distribution contractions/expansions of the mole shrews probably repeated many times with different refugial sizes and durations over the last 5.4 myr (Stewart et al., 2010). The true scenario could be very complicated and resulted in the extinction of the all mole shrew species but one.
The SDM also suggested that the southern subregion of MSC have relatively more stable climatic conditions. This result explains the higher genetic diversity preserved in this area and supported by the continuous-diffusion phylogeographic analyses. The distributions of clades S1, S2 and S4, which marginally surround the southern Hengduan mountains, imply existence of microrefugia in these outlier areas (Rull, 2009). Although we could not determine the precise locations of microrefugia, because of the insufficient precision and accuracy of the projected climate data and/or insufficient sampling from these areas, our results suggested microrefugia existed in northern Myanmar, Vietnam and Thailand.
Interestingly, the southern subregion of the Hengduan Mountain also harbors higher species richness of seed plants compared with the northern subregion, which was originally explained as a result of the greater extremes of topography and wider diversity of habitats (Zhang et al., 2009). Our results suggested that more stable habitats and likely more microrefugia may be other reasons for the high species diversity in the southern subregion of the mountains.
Conclusions
This is the first study to show to which degree the topography and climate affected the genetic diversity of a small mammal species in China. The topography did not have strong influence in shaping the phylogenetic structure of A. squamipes. In contrast, climate fluctuations strongly affected the distribution, migration, demography of the species and are likely the cause of the extinction of fossil species. A. squamipes could have retreated to refugia during unfavorable periods (the interglaciations) because of greater climatological seasonality and expanded and/or colonized new habitats during the last glaciations. The southern subregion of the MSC provided larger stable habitats, which allowed the survival of mole shrews in a macrorefugium and multiple microrefugia and preserved higher genetic diversity.
Data archiving
Sequences for voucher specimens were submitted to GenBank under accession numbers KT032255–KT032564 for ND2 sequences and KT032565–KT032946 for CYT B sequences.
Accession codes
References
Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV . (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol 29: 2157–2167.
Baldwin RA . (2009). Use of maximum entropy modeling in wildlife research. Entropy 11: 854–866.
Bennett KD, Provan J . (2008). What do we mean by 'refugia'? Quat Sci Rev 27: 2449–2455.
Bielejec F, Rambaut A, Suchard MA, Lemey P . (2011). SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 27: 2910–2912.
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R et al. (2014). DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30: 1187–1189.
Drummond AJ, Suchard MA, Xie D, Rambaut A . (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973.
Edgar RC . (2014). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
He K, Jiang X . (2014). Sky islands of southwest China. I: An overview of phylogeographic patterns. Chin Sci Bull 59: 585–597.
He K, Li YJ, Brandley MC, Lin LK, Wang YX, Zhang YP et al. (2010). A multi-locus phylogeny of Nectogalini shrews and influences of the paleoclimate on speciation and evolution. Mol Phylogenet Evol 56: 734–746.
Heled J, Drummond AJ . (2008). Bayesian inference of population size history from multiple loci. BMC Evol Biol 8: 1–15.
Hewitt G . (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc (UK) 58: 247–276.
Hewitt G . (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond Ser B 359: 183–195.
Ho SYW, Lanfear R . (2010). Improved characterisation of among-lineage rate variation in cetacean mitogenomes using codon-partitioned relaxed clocks. Mitochondrial DNA 21: 138–146.
Huang PH, Zheng LZ, Quan YC . (1995). Preliminary Study on ESR dating of Hexian-Man and its fauna. Nucl Techniques 18: 491–494.
Lanfear R, Calcott B, Ho SYW, Guindon S . (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29: 1695–1701.
Lemey P, Rambaut A, Welch JJ, Suchard MA . (2010). Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol 27: 1877–1885.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Liu Q, Chen P, He K, Kilpatrick CW, Liu S-Y, Yu F-H et al. (2012). Phylogeographic study of Apodemus ilex (Rodentia: Muridae) in Southwest China. PLoS One 7: e31453.
Louis M, Fontaine MC, Spitz J, Schlund E, Dabin W, Deaville R et al. (2014). Ecological opportunities and specializations shaped genetic divergence in a highly mobile marine top predator. Proc R Soc Ser B 281: 1795.
Merritt JF . (1986). Winter survival adaptations of the short-tailed shrew (Blarina brevicauda) in an Appalachian montane forest. J Mammal 67: 450–464.
Ohdachi SD, Hasegawa M, Iwasa MA, Vogel P, Oshida T, Lin LK et al. (2006). Molecular phylogenetics of soricid shrews (Mammalia) based on mitochondrial cytochrome b gene sequences: with special reference to the Soricinae. J Zool 270: 177–191.
Otto-Bliesner BL, Marsha SJ, Overpeck JT, Miller GH, Hu AX, CLIP Mem . (2006). Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science 311: 1751–1753.
Paradis E, Claude J, Strimmer K . (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289.
Phillips SJ, Dudik M . (2008). Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31: 161–175.
Rogers A, Harpending H . (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9: 552–569.
Rull V . (2009). Microrefugia. J Biogeogr 36: 481–484.
Stamatakis A, Hoover P, Rougemont J . (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57: 758–771.
Stewart JR, Lister AM, Barnes I, Dalén L . (2010). Refugia revisited: individualistic responses of species in space and time. Proc R Soc Ser B 277: 661.
Storch G, Qiu Z, Zazhigin VS . (1998) Fossil history of shrews in Asia. In: Wojcik JM, Wolsan M (eds). Evolution of Shrews. Mammal Research Insitute: Bialowieza, Poland, pp 93–120.
Sun YB, Clemens SC, An ZS, Yu ZW . (2006). Astronomical timescale and palaeoclimatic implication of stacked 3.6- Myr monsoon records from the Chinese Loess Plateau. Quat Sci Rev 25: 33–48.
van Dam JA . (2004). Anourosoricini (Mammalia: Soricidae) from the Mediterranean region: a pre-quaternary example of recurrent climate-controlled north-south range shifting. J Paleontol 78: 741–764.
Wang WJ, McKay BD, Dai CY, Zhao N, Zhang RY, Qu YH et al. (2013). Glacial expansion and diversification of an East Asian montane bird, the green-backed tit (Parus monticolus. J Biogeogr 40: 1156–1169.
Wiens JJ, Graham CH . (2005). Niche conservatism: integrating evolution, ecology, and conservation biology. Annu Rev Ecol Evol Syst 36: 519–539.
Wojcik JM, Wolsan M . (1998) Evolution of Shrews. Mammal Research Institute: Bialowieza, Poland.
Yang Z, Rannala B . (2010). Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci USA 107: 9264.
Yuan S-L, Jiang X-L, Li Z-J, He K, Harada M, Oshida T et al. (2013). A mitochondrial phylogeny and biogeographical scenario for asiatic water shrews of the genus Chimarrogale: Implications for taxonomy and low-latitude migration routes. PLoS One 8: e77156.
Yuan SL, Lin LK, Oshida T . (2006). Phylogeography of the mole-shrew (Anourosorex yamashinai in Taiwan: implications of interglacial refugia in a high-elevation small mammal. Mol Ecol 15: 2119–2130.
Zhang DC, Boufford DE, Ree RH, Sun H . (2009). The 29°N latitudinal line: an important division in the Hengduan mountains, a biodiversity hotspot in southwest China. Nord J Bot 27: 405–412.
Zhang RZ . (2002). Geological events and mammalian distribution in China. Acta Zool Sin 48: 141–153.
Ziegler R . (1989). Heterosoricidae und Soricidae (Insectivora, Mammalia) aus dem Oberoligozän und Untermiozän Süddeutschlands. Stuttgarter Beitr Naturk B154: 1–73.
Acknowledgements
We appreciate the constructive comments and suggestions from the two anonymous reviewers. We thank Dr Shaoying Liu, Dr Shunde Chen and Dr Qingyong Ni for providing us samples from Sichuan. We also thank Dr Filip Bielejec for providing us the newest version of SPREAD, Dr Philippe Lemey and Dr Arley Camargo for providing code for constructing dispersal rate through time plot and Dr Youbin Sun and Mr Peter Shum for their very useful discussions. We are grateful to Dr Shouli Yuan for many constructive discussions. This paper is modified from Chapter 6 of the PhD thesis of KH. The study is supported by the Key Research Program of the Chinese Academy of Sciences (KJZD-EW-L07) and the National Natural Science Foundation of China (31301869).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
He, K., Hu, NQ., Chen, X. et al. Interglacial refugia preserved high genetic diversity of the Chinese mole shrew in the mountains of southwest China. Heredity 116, 23–32 (2016). https://doi.org/10.1038/hdy.2015.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.62
This article is cited by
-
The chloroplast genomes of four Bupleurum (Apiaceae) species endemic to Southwestern China, a diversity center of the genus, as well as their evolutionary implications and phylogenetic inferences
BMC Genomics (2021)
-
Coalescence Models Reveal the Rise of the White-Bellied Rat (Niviventer confucianus) Following the Loss of Asian Megafauna
Journal of Mammalian Evolution (2019)
-
Disjunct distribution and distinct intraspecific diversification of Eothenomys melanogaster in South China
BMC Evolutionary Biology (2018)
-
An endemic rat species complex is evidence of moderate environmental changes in the terrestrial biodiversity centre of China through the late Quaternary
Scientific Reports (2017)
-
The roles of barriers, refugia, and chromosomal clines underlying diversification in Atlantic Forest social wasps
Scientific Reports (2017)
