
Abstract
Given predicted rapid climate change, an understanding of how environmental factors affect genetic diversity in natural populations is important. Future selection pressures are inherently unpredictable, so forest management policies should maintain both overall diversity and identify genetic markers associated with the environmental factors expected to change most rapidly, like temperature and rainfall. In this study, we genotyped 648 individuals in 28 populations of Castanopsis fargesii (Fagaceae) using 32 expressed sequence tag (EST)-derived microsatellite markers. After removing six loci that departed from Hardy–Weinberg equilibrium, we measured genetic variation, population structure and identified candidate loci putatively under selection by temperature and precipitation. We found that C. fargesii populations possessed high genetic diversity and moderate differentiation among them, indicating predominant outcrossing and few restrictions to gene flow. These patterns reduce the possible impact of stochastic effects or the influence of genetic isolation. Clear footprints of divergent selection at four loci were discovered. Frequencies of five alleles at these loci were strongly correlated with environmental factors, particularly extremes in precipitation. These alleles varied from being near fixation at one end of the gradient to being completely absent at the other. Our study species is an important forest tree in the subtropical regions of China and could have a major role in future management and reforestation plans. Our results demonstrate that the gene flow is widespread and abundant in natural populations, maintaining high diversity, while diversifying selection is acting on specific genomic regions.
Similar content being viewed by others

Introduction
Both global and local patterns of precipitation and temperature are rapidly changing (IPCC, 2007), altering community composition and shifting range boundaries, phenology, genetic diversity and genetic structure of organisms (Walther et al., 2002; Manel et al., 2012). Plants can respond to global climate change through several mechanisms, including range shift migration, phenotypic plasticity and local adaptation (Aitken et al., 2008; Holderegger et al., 2008). However, simple migration will probably not be sufficient to allow populations to track their current optimal environment (Jump and Peñuelas, 2005). In addition, while most plants do exhibit some level of phenotypic plasticity, the range of responses will probably not be adequate to cope with local climate change (Anderson et al., 2012). Therefore, local adaptation to future climatic conditions will have an important role for the long-term in situ persistence of native plant populations (Holderegger et al., 2008; Anderson et al., 2012).
Detecting adaptive genetic variation in response to climatic variation is a compelling and challenging task, particularly in non-model plants for which genomic information is still limited or absent (Manel et al., 2012). Expressed sequence tag (EST) databases have been collected for many plant species, and can be mined to generate markers that are tightly associated with gene-rich regions and particularly useful for exploring the genome for possible signatures of selection (Namroud et al., 2008). Gene loci that show unusually low or high levels of genetic differentiation (FST) are often assumed to be subject to natural selection (Beaumont and Nichols, 1996; Storz, 2005; Antao et al., 2008; Excoffier et al., 2009). An increasing number of studies have been carried out to detect FST outlier loci by exploring a larger sampling of distinct populations and markers (Eveno et al., 2008). In addition, testing genetic-environmental correlations across genomic data sets is often used to detect signatures of natural selection (Joost et al., 2007; Holderegger et al., 2008).
Forest trees are ideal for studying the genetic basis of local adaptation because of their large open-pollinated native populations, abundant genetic and phenotypic variation, low genetic differentiation among populations at neutral genetic markers and much higher levels of latitudinal or altitudinal differentiation in adaptive traits (Savolainen et al., 2007). Identifying genes that are under selection in natural populations of tree species will facilitate our understanding of how these populations are adapted to their environment and responded to future climatic changes (Bradbury et al., 2013). Evidence for selection at the molecular genetic level has been demonstrated in some important tree species such as spruce (Namroud et al., 2008), pine (Eveno et al., 2008), oak (Alberto et al., 2013) and Eucalyptus (Bradbury et al., 2013).
Castanopsis fargesii (Fagaceae) is an evergreen broad-leaved tree species that dominates in forests of subtropical China. For plants in this region, many phylogeographic studies suggest a congruent pattern of historical fragmentation and only localized or no range expansions (Liu et al., 2013), yet few of them focus on divergent selection or local adaptation. Subtropical China is marked by clear latitudinal temperature (cooler in the north) and longitudinal precipitation (wetter in the east) gradients. These environmental gradients can impose strong selective pressure on plant populations, driving local adaptation (Jump et al., 2006; Eveno et al., 2008). Morphological variation on leaves and cupules of C. fargesii are observed across its whole distribution range (Huang and Chang, 1998), and a high level of genetic diversity of four populations of C. fargesii in Fujian province has been detected using six genomic microsatellite molecular markers (Xu et al., 2001). Leaf mass per area of this species negatively correlates with mean annual temperature and mean annual precipitation (Dong et al., 2009). Based on these observations of putatively adaptive phenotypic traits, spatially varying environmental selection is expected to influence the distribution of genetic variation across populations in this species.
Here, we expand the population and genomic sampling effort for this species to include 32 EST-derived simple sequence repeat (SSR) markers in 28 populations distributed across the majority of the natural range of C. fargesii in the evergreen broad-leaved forests of subtropical China. We specifically intend to (1) detect the existing genetic variation and population structure of this dominant tree species in the remaining evergreen broad-leaved forests of subtropical China; (2) identify non-neutral genetic outlier putatively under divergent selection; and (3) test association of allelic frequencies at candidate outlier loci with climatic variation.
Materials and methods
Samples, DNA extraction and EST-SSR genotyping
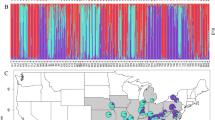
A total of 28 populations comprising 648 individuals of C. fargesii were collected across the species range in the mainland of China (Figure 1; Table 1). Voucher specimens were made for each population and stored in the Herbarium of the South China Botanical Garden (formerly the South China Institute of Botany, IBSC), Chinese Academy of Science. Sampled trees were at least 20 m apart to avoid sampling of close relatives. Fresh leaves were stored in silica gel before DNA extraction.

Localities and geographical pattern of climatic variation of 28 populations of Castanopsis fargesii sampled in China. Circles represent localities, and colors represent site PC1 scores based on principal coordinates analysis of eight climatic variables including isothermality, temperature seasonality, maximum temperature of warmest month, temperature annual range, mean temperature of warmest quarter, precipitation in driest month, driest quarter and coldest quarter.
Total genomic DNA was extracted following cetyltrimethyl ammonium bromide protocol described in Doyle and Doyle (1987). PCR was performed in a total volume of 10 μl, which contained 1.5 mM of MgCl2, 0.2 μM of each forward and reverse primer, 0.2 mM each of dNTPs, 0.4 unit of Taq DNA polymerase (TaKaRa, Dalian, China), 1 μl of 10 × PCR buffer and about 20 ng of DNA template. PCR was conducted on a SensoQuest labcycler (SensoQuest, Göttingen, Germany) using the following program: an initial pre-denaturation at 94 °C for 3 min, followed by 35 cycles including 45 s at 94 °C, 45 s at 50–60 °C and 45 s at 72 °C and a final extension step at 72 °C for 7 min. Primers were labeled at 5′-end with the fluorochromes HEX, 6-FAM, TAMRA and ROX. PCR products were visualized on an ABI-377 fluorescence sequencer (Applied Biosystems, Carlsbad, CA, USA). Alleles were scored with GeneMapper version 1.51 (Applied Biosystems).
A total of 168 EST-SSR primer pairs, including 124 polymorphic EST-SSRs in Castanea mollissima (www.fagaceae.org), 16 in Castanopsis sieboldii (Ueno et al., 2009) and 28 in Quercus mongolica (Ueno et al., 2008; Ueno and Tsumura, 2008), were screened in a pilot experiment using 20 individuals of C. fargesii. Finally, 32 primer pairs (Table 2) with clear, polymorphic banding patterns were picked to genotype all individuals.
Climate data
Nineteen climatic variables on temperature and precipitation during the period from 1950 to 2000 at the sampled locations were obtained by interpolating climate layers from a standard set of climate grids (http://www.worldclim.org/) with a spatial resolution of about one square kilometer (Hijmans et al., 2005). Principal coordinates analysis were performed using PRIMER v6 (Clarke and Gorley, 2006) to get the principal components that best summarized the overall pattern of variation in these variables.
Data analyses
The number of alleles observed (NA), allelic richness (AR) rarefied to the smallest sample size of 10 diploid individuals per population, observed heterozygosity (HO), gene diversity within population (HS), gene diversity in the total population (HT), genetic differentiation among populations (FST) and inbreeding coefficient (FIS) were estimated by using FSTAT 2.9.3 (Goudet, 2001). Genotypic disequilibrium between all pairs of loci and Hardy–Weinberg equilibrium were tested for each population; P-values were subject to Bonferroni correction for multiple comparisons. The frequencies of null alleles were estimated for each locus by using the program FreeNA (Chapuis and Estoup, 2007). Six loci that significantly departed from Hardy–Weinberg equilibrium were removed in further analyses of genetic structure, outlier test and spatial correlation association.
Genetic structure was assessed by using only those markers that showed no indication of outlier behavior in LOSITAN analysis (see below) with a model-based Bayesian clustering method implemented in the program STRUCTURE version 2.2 (Pritchard et al., 2000). The analyses were conducted with the admixture model and the option of correlated allele frequencies between populations. The length of burn-in and Markov chain Monte Carlo were set to 30 000 and 100 000, respectively. Five replicated runs were carried out for each possible number of clusters (K) being tested from 1 to 28. The statistic of ΔK, based on the second order rate of change of L(K) between successive K values, was calculated by STRUCTURE HARVESTER (Earl and vonHoldt, 2012) to identify the most appropriate number of clusters. Using neutral markers, analysis of molecular variance was conducted with ARLEQUIN 3.5 (Excoffier and Lischer, 2010) to partition genetic variance among populations and within populations.
To increase the accuracy of candidate loci detection in this study, a number of methods, as many populations as possible and correction of allele frequency autocorrelation were employed to detect selection footprint according to the suggestion of a research based on simulation data (De Mita et al., 2013). At first, the FST outlier approach (Beaumont and Nichols, 1996) implemented in LOSITAN (Antao et al., 2008) was used to identify outlier loci. Outlier values of FST were identified from a plot of FST vs heterozygosity that was generated under a simple island model. In LOSITAN analysis, 100 000 simulations were run by using the stepwise mutation model with the option of neutral mean FST to generate the expected distributions of FST vs heterozygosity. Markers that presented FST higher than the 95% confidence interval were considered candidates for divergent selection, and markers that presented FST lower than the 95% confidence interval were considered candidates for balancing selection. False discovery rate in LOSITAN analysis was set to 0.01. Second, the hierarchical structure among populations revealed by the results of the clustering was taken into account to avoid possible false positives. The hierarchical island model implemented in ARLEQUIN 3.5 (Excoffier and Lischer, 2010) was used to obtain more realistic null distribution for FST statistics. Each population was assigned to a group based on the average proportion of membership (Q) calculated from STRUCTURE analysis with neutral markers. Populations were assumed as admixed if Q value was <0.68. These populations were removed before outlier testing by ARLEQUIN. Coalescent simulations (500 000) were performed under a hierarchical island model to get the null distribution of F-statistics.
Association between allelic frequencies at marker loci and environmental variables were tested with multiple univariate logistic regressions by using spatial analysis method (SAM) (Joost et al., 2007). The likelihood ratio (G) and Wald tests were used to determine the significance of the models. Statistical signals were considered significant at 99.9999999999% confidence level by applying the Bonferroni correction for multiple comparisons. To correct allele frequency autocorrelation, significant associations in SAM were re-evaluated using univariate linear regressions between population allele frequencies and environmental variables (Narum et al., 2010).
Results
Genetic diversity and structure
A total of 370 alleles were revealed at 32 loci. The number of alleles observed (NA) varied from 4 to 21 alleles per locus (Table 2), and allelic richness (AR) at different loci ranged from 2.035 to 8.686. No significant departures from linkage equilibrium were detected for any pair of loci in each population. Six loci significantly (P<0.01) deviated from Hardy–Weinberg equilibrium after Bonferroni correction. The estimated null allele frequency at the six loci varied from 0.043 to 0.131. The six non-Hardy–Weinberg equilibrium loci were removed in further analyses.
C. fargesii possessed high putatively neutral diversity. A total of 205 alleles were revealed at 18 neutral loci (see LOSITAN results). Among populations, the number of alleles observed and allelic richness ranged from 4 to 6 and 3.735 to 5.592, respectively. Gene diversity in each population varied from 0.568 to 0.708 with an average value of 0.655. Overall inbreeding coefficient and genetic differentiation was 0.006 and 0.105, respectively.
Hierarchical analysis of molecular variance of 18 neutral markers showed that 89.52% of the total genetic variation occurred within populations, and 10.48% among populations. Population genetic structure was revealed using STRUCTURE analysis with 18 neutral markers. The plot of ΔK against a range of K values (from 1 to 28) revealed a distinct peak at K=2. In general, two core genetic groups were evident, representing a western group (Yingjing to Yuelushan) vs an eastern group (Jinggangshan to Ningbo) (Figure 2). High (above 68%) cluster membership was observed in the west (Yingjing to Yuelushan, with the exception of Malipo) and east (Jinggangshan to Ningbo, with the exception of Gutianshan), with admixture in populations of Dagangshan and Lushan.

Genetic structure of Castanopsis fargesii and individual proportion of the membership for the inferred clusters defined by the STRUCTURE analysis using neutral loci.
Genetic outlier analyses
In LOSITAN analysis, seven loci (CC2091, CC2448, CC3754, CC4323, CC5223, CC17354 and CcC00660) were identified possibly under divergent selection with a confidence level of 95%, while CC20303 was putatively under balancing selection (Figure 3). After taking population structure into account, ARLEQUIN analyses identify three loci (CC2091, CC2448 and CC5223) putatively under divergent selection with a confidence level of 95%, while three loci (CC20303, CcC02075 and CcC02464) were under balancing selection (Figure 4).

Outlier loci detected with LOSITAN software. Outlier loci were significant at the 5% level.

Outlier loci detected with ARLEQUIN software. Outlier loci were significant at the 5% level.
Genetic variation associated with climate variables
In SAM analysis, eight climatic variables including isothermality, temperature seasonality, maximum temperature of warmest month, temperature annual range, mean temperature of warmest quarter, precipitation in driest month, driest quarter and coldest quarter were revealed as significantly associated with allelic frequencies (Table 3). The overall trend of these eight climatic variables was captured by principal components analysis, and the first principal component (PC1) explained 70.7% of the total variation. The geographical pattern of the climatic gradients is shown in Figure 1, site PC1 scores are generally high in east and low in west, depicting an obvious climatic gradient along southwest to northeast in sampled area (Figure 1).
Four loci (CC2091, CC2448, CC3754 and CcC00087) exhibited significant association with one or more climate factors by both G and Wald tests with a significance threshold set to 1.93E−16, corresponding to a 99.9999999999% confidence level including Bonferroni correction. Precipitation of coldest quarter was the most often climate variable detected in these significant allele-environment associations.
Allele 175 bp at locus CC2091 (CC2091-175) was significantly associated with maximum temperature of warmest month, mean temperature of warmest quarter and precipitation of coldest quarter (Table 3; Figure 5a). Allele 174 bp at locus CC2448 (CC2448-174) showed significant association with precipitation of coldest quarter (Table 3; Figure 5b). Two alleles from CC3754 exhibited a total of eight significant allele–environment associations—five climate variables including maximum temperature of warmest month, mean temperature of warmest quarter, and precipitation of driest month, driest quarter and coldest quarter associated with allele CC3754-307 (Table 3; Figure 5c), while isothermality, temperature seasonality and temperature annual range associated with allele CC3754-310 (Table 3; Figure 5d). Allele 388 bp at locus CcC00087 (CcC00087-388) was significantly associated with precipitation of coldest quarter and driest month (Table 3; Figures 5e and f).

Correlations between climatic variation and allele frequencies at loci (a) CC2091; (b) CC2448; (c) and (d) CC3754; (e) and (f) CcC00087.
Discussion
In this study, we explored EST-SSR diversity in 648 individuals of C. fargesii sampled from 28 natural populations distributed across strong environmental gradients of temperature and precipitation in central to southern China. We identified strong signatures of diversifying selection at four loci (CC2091, CC2448, CC3754 and CC5223). Given the predicted rapid change in global climate, the response of natural forest populations will depend on the genetic diversity and spatial distribution of alleles for these genomic regions that are under clear selection pressure. Our results provide useful information on both neutral and adaptive genetic diversity of C. fargesii and its spatial distribution in relation to key environmental factors. We discovered informative genetic markers related to the future evolution, adaptation and management of this major forest species in the subtropics of Asia. The sampling and analytical approach pursued here could provide general insights into the adaptive responses of plant species to future climatic change.
Natural populations of C. fargesii are predominantly out-crossed with little indication of geographic restrictions in gene flow, high overall levels of genetic diversity (H=0.655) and little evidence of inbreeding (FIS=0.006). Castanopsis species should have high potential for long-distance dispersal, particularly for pollen, given the fat-tailed dispersal kernel observed by Nakanishi et al., (2012). Long-distance dispersal events have strong impact on population structure and adaptation (Savolainen et al., 2007), and along with sexual outcrossing, allow alleles to move easily among populations, since population differentiation was only moderate (FST=0.105). The spatial pattern that emerges for a specific locus can then be assumed to be more closely related to selective pressures instead of random genetic drift or stochastic isolation-by-distance processes. Generally, high levels of genetic diversity and unrestricted gene flow within a species are crucial for adaptation, especially when facing unpredictable local environmental changes.
The range-wide geographic mosaic distributions of lineages caused by long-term fragmentation are often observed in plants in subtropical China (Liu et al., 2013). Population differentiation of C. fargesii is revealed by neutral markers in STRUCTURE analysis; the 28 studied populations of C. fargesii are clustered into two core genetic groups, representing a western group (Yingjing to Yuelushan) vs an eastern group (Jinggangshan to Ningbo). The differentiation between western and eastern groups observed in C. fargesii is most likely a result of isolation. Roughly along the populations of Tianjinshan, Chebaling, Jiulians, Jinggangshan, Jianning, Wuyishan and Gutianshan, there are several major mountain ranges situated including Nanling mountain, Jingang mountain, Wuyi mountain and Gutian mountain, may act as a physical barrier to reduce gene exchange between two groups or gene pools. Neutral divergence of two groups was low although significant; analysis of molecular variance analysis revealed that only 2.63% of the total genetic variance resided among groups, suggesting extensive gene flow among groups.
The distribution pattern of Castanopsis species are strongly affected by environment (Liu and Hong, 1998). The geographic patterns of climatically structured genetic variation provided a useful indication of the geographic pattern of adaptive variation (Sork et al., 2010). An obvious climatic gradient presented along southwest to northeast in sampled area (Figure 1), especially for precipitations in driest month, driest quarter and coldest quarter, these variables are generally low in the western portion of the sampled area, increasing to the east (Table 1). Given that strong signatures of diversifying selection at four outliers and their allelic frequencies were directly correlated with climate variables, existing climatic gradients obviously have a large impact on the genetic structure and diversity of C. fargesii. High gene flow has not prevented rapid adaptive divergence of extant populations of C. fargesii along environmental gradients.
Climatic factors, particularly precipitation and temperature, were important driving forces in population differentiation along longitudinal and latitudinal clines in different plant species (Savolainen et al., 2007; Manel et al., 2012). Local adaptation of plant populations was one of the most effective mechanisms to cope with rapid climate change, particularly if migration capacity was hampered by a highly modified and fragmented landscape and response through phenotypic plasticity was insufficient (Manel et al., 2012). While most of the EST-SSRs in this study were presumably neutral markers, we found that four loci (CC2091, CC2448, CC3754 and CC5223) appeared to be under diversifying selection because they were detected by at least two methods. Three programs including LOSITAN, Arlequin and SAM were performed to find genetic footprint of divergent selection. Three loci (CC2091, CC2448 and CC5223) were identified as FST outlier putatively under divergent selection by both LOSITAN and Arlequin analysis. Locus CC3754 was revealed as candidate loci under divergent selection by LOSITAN and SAM analysis. The frequencies of allele 307 bp at CC3754 and allele 175 bp at CC2091 were significantly correlated with multiple variables of both precipitation and temperature, agreeing with previous results indicating that cold and drought tolerance was physiologically correlated in oak species (Gimeno et al., 2009), and that precipitation and temperature were key factors generally determining the geographic distribution of Castanopsis species (Liu and Hong, 1998).
The locus CC2448 was functionally assigned to Poly-ADP-ribose transferase. Radical-induced cell death1 (RCD1) and proteins similar to RCD1, which contained the core catalytic domain of poly-ADP-ribose transferase, were important regulators of stress and developmental responses in plants (Jaspers et al., 2009). Our results suggested that this locus might be involved in climatic adaptation of C. Fargesii, especially associated with climatic extremes such as precipitation of driest and coldest quarter. Gene ontology function of CC5223 was assigned to blue copper-binding protein. The expression of blue copper-binding protein was found to be upregulated during drought stress in Arabidopsis (Seki et al., 2002). Blue copper-binding proteins were also functionally expressed in fiber (epidermal trichome) development in cotton (Ruan et al., 2011). Potentially related morphological variation on leaves and cupules of C. fargesii has been observed. The underside of the leaves were covered by scalelike trichomes, which were thick and mealy in southeastern China but thin and appressed in southwestern provinces such as Sichuan and Guizhou (Huang and Chang, 1998). Leaf mass per area of C. fargesii was shown to be negatively correlated with mean annual temperature and precipitation (Dong et al., 2009). Leaf trait associated with high leaf mass per area had been interpreted as an adaptation under very dry conditions in evergreen species (Wright et al., 2005). Given these patterns, blue copper-binding proteins might be involved in trichome production and drought tolerance of C. fargesii, providing a potential avenue for further detailed investigation. The molecular functions of CC2091 and CC3754 were not discovered. Three loci (CC20303, CcC02075 and CcC02464) was identified as possibly being under balancing selection. The first two loci were linked to uncharacterized protein, while CcC02464 assigned to a 5′-nucleotidase SurE-like survival protein in Arabidopsis (Ueno et al., 2009).
In the face of uncertain but potentially rapid climate change, two aspects of population level genetic diversity should be the focus of sound management and reforestation practices in the subtropics of China. First, high levels of genetic diversity and connectivity should be maintained in all populations to facilitate natural adaptation to unpredictable climate shifts. As few boundaries to gene flow were observed in this genetically diverse species, foresters should monitor these levels of diversity to ensure connectivity persists, especially between the eastern and western groups revealed in this study, and genetic mixture continues so that appropriate genetic combinations for future climate changes can easily arise. Second, reforestation schemes should screen seedling stock to enrich appropriate genetic markers in regenerating forest and prevent the use of mother trees possessing disadvantageous genes. Particularly, if assisted gene flow approaches are utilized to increase dispersal and facilitate local adaptation to climate change (Aitken and Whitlock, 2013), the genetic markers identified here can be used to artificially select individuals with appropriate alleles for transplanting, thus enhancing the ability of populations to adapt and respond.
In general, the genus Castanopsis was an important tree group in the management of subtropical forests in China, being a major component of reforestation and management efforts. The results of this study, particularly the discovery of genetic markers directly associated with clear environmental gradients, and extremes in precipitation, will assist in the effective artificial selection and screening of stock material for transplanting and reforestation programs across a heterogeneous and dynamic and unpredictable landscape.
Data archiving
Data available from the Dryad Digital Repository: 10.5061/dryad.hf49c.
References
Aitken SN, Whitlock MC . (2013). Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44: 367–388.
Aitken SN, Yeaman S, Holliday JA, Wang TL, Curtis-McLane S . (2008). Adaptation, migration or extirpation: climate change outcomes for tree population. Evol Appl 1: 95–111.
Alberto FJ, Derory J, Boury C, Frigerio JM, Zimmermann E, Kremer A . (2013). Imprints of natural selection along environmental gradients in phenology-related genes of Quercus petraea. Genetics 195: 495–512.
Anderson JT, Inouye DW, McKinney AM, Colautti RI, Mitchell-Olds T . (2012). Phenotypic plasticity and adaptive evolution contribute to advancing flowering phenology in response to climate change. Proc R Soc B Bio Sci 279: 3843–3852.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G . (2008). LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9: 323.
Beaumont MA, Nichols RA . (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc B Bio Sci 263: 1619–1626.
Bradbury D, Smithson A, Krauss SL . (2013). Signatures of diversifying selection at EST-SSR loci and association with climate in natural Eucalyptus populations. Mol Ecol 22: 5112–5129.
Chapuis MP, Estoup A . (2007). Microsatellite null alleles and estimation of population differentiation. Mol Bio Evol 24: 621–631.
Clarke KR, Gorley RN . (2006) PRIMER v6: User Manual/Tutorial. PRIMER-E: Plymouth, MA, USA.
De Mita S, Thuillet A, Gay L, Ahmadi N, Manel S, Ronfort J et al. (2013). Detection selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol Ecol 22: 1383–1399.
Dong LL, Liu SR, Shi ZM, Feng WH . (2009). Relationships between leaf traits of Castanopsis species and the environmental factors in the north-south transect of eastern China. For Res 22: 463–469.
Doyle JJ, Doyle JL . (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 1–3.
Eveno E, Collada C, Guevara MA, Léger V, Soto A, Díaz L et al. (2008). Contrasting patterns of selection at Pinus pinaster Ait. drought stress candidate genes as revealed by genetic differentiation analysis. Mol Bio Evol 25: 417–437.
Excoffier L, Hofer T, Foll M . (2009). Detecting loci under selection in a hierarchically structured population. Heredity 103: 285–298.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Gimeno TE, Pias B, Lemos-Filho JP, Valladares F . (2009). Plasticity and stress tolerance override local adaptation in the responses of Mediterranean holm oak seedlings to drought and cold. Tree Physiol 29: 87–98.
Goudet J . (2001) FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9. 3). Available at http://www2.unil.ch/popgen/softwares/fstat.htm.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A . (2005). Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25: 1965–1978.
Holderegger R, Herrmann D, Poncet B, Gugerli F, Thuiller W, Taberlet P et al. (2008). Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Divers 1: 273–283.
Huang CJ, Chang YT . (1998). Castanopsis. In: Chun WY, Huang CJ, (eds) Flora Reipublicae Popularis Sinicae vol. 22, Science Press: Beijing, China. pp 13–80.
IPCC. (2007). Climate change 2007: the physical science basis. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds) Contribution of Working Group to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press: Cambridge, England.
Jaspers P, Blomster T, Brosché M, Salojärvi J, Ahlfors R, Vainonen JP et al. (2009). Unequally redundant RCD1 and SRO1 mediate stress and developmental responses and interact with transcription factors. Plant J 60: 268–279.
Joost S, Bonin A, Bruford M, Després L, Conord C, Erhardt G et al. (2007). A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16: 3955–3969.
Jump AS, Hunt JM, Martinez-Izquierdo JA, Peñuelas J . (2006). Natural selection and climate change: temperature-linked spatial and temporal trends in gene frequency in Fagus sylvatica. Mol Ecol 15: 3469–3480.
Jump AS, Peñuelas J . (2005). Running to stand still: adaptation and the response of plant to rapid climate change. Ecol Lett 8: 1010–1020.
Liu MS, Hong BG . (1998). The distribution of Fagaceae in China and its relationship with climatic and geographic characters. Acta Phytoecol Sin 22: 41–50.
Liu ZY, Wu R, Wang Q, Zhang ZR, Pujol JL, Fan DM et al. (2013). Comparative phylogeography of two sympatric beeches in subtropical China: species-specific geographic mosaic of lineages. Ecol Evol 3: 4461–4472.
Manel S, Gugerli F, Thuiller W, Alvarez N, Legendre P, Holderegger R et al. (2012). Broad-scale adaptive genetic variation in alpine plants is driven by temperature and precipitation. Mol Ecol 21: 3729–3738.
Nakanishi A, Yoshimaru H, Tomaru N, Miura M, Manabe T, Yamamoto SI . (2012). Patterns of pollen flow in a dense population of the insect-pollinated canopy tree species Castanopsis sieboldii. J Hered 103: 547–556.
Namroud MC, Beaulieu J, Juge N, Laroche J, Bousquet J . (2008). Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol Ecol 17: 3599–3613.
Narum SR, Campbell NR, Kozfkay CC, Meyer KA . (2010). Adaptation of redband trout in desert and montane environments. Mol Ecol 19: 4622–4637.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Ruan XM, Luo F, Li DD, Zhang J, Liu ZH, Xu WL et al. (2011). Cotton BCP genes encoding putative blue copper-binding proteins are functionally expressed in fiber development and involved in response to high-salinity and heavy metal stress. Physiol Plant 141: 71–83.
Savolainen O, Pyhäjärvi T, Knürr T . (2007). Gene flow and local adaptation in trees. Annu Rev Ecol Evol Syst 38: 595–619.
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y et al. (2002). Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarry. Plant J 31: 279–292.
Sork VL, Davis FW, Westfall R, Flint A, Ikegami M, Wang HF et al. (2010). Gene movement and genetic association with regional climate gradients in California valley oak (Quercus lobata Nee) in the face of climate change. Mol Ecol 19: 3806–3823.
Storz JF . (2005). Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol Ecol 14: 671–688.
Ueno S, Aoki K, Tsumura Y . (2009). Generation of expressed sequence tags and development of microsatellite markers for Castanopsis sieboldii var. sieboldii (Fagaceae). Ann For Sci 66: 509–509.
Ueno S, Taguchi Y, Tsumura Y . (2008). Microsatellite markers derived from Quercus mongolica var. crispula (Fagaceae) inner bark expressed sequence tags. Genes Genet Syst 83: 179–187.
Ueno S, Tsumura Y . (2008). Development of ten microsatellite markers for Quercus mongolica var. crispula by database mining. Conserv Genet 9: 1083–1085.
Walther GR, Post E, Convey P, Menzel A, Parmesan C, Beebee TJC et al. (2002). Ecological responses to recent climate change. Nature 416: 389–395.
Wright IJ, Reich PB, Cornelissen JHC, Falster DS, Groom PK, Hikosaka K et al. (2005). Modulation of leaf economic traits and trait relationships by climate. Global Ecol Biogeogr 14: 411–421.
Xu LA, Li XJ, Pan HX, Zou HY, Yin TM, Huang MR . (2001). Study on population genetic structure in Castanopsis fargesii with microsatellite markers. Acta Bot Sin 43: 409–412.
Acknowledgements
We thank anonymous reviewers for their critical comments on an earlier version of the manuscript. This study was financially supported by the National Natural Science Foundation of China (30871959, 31170512 and 31370668) and the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-J-28).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, C., Sun, Y., Huang, H. et al. Footprints of divergent selection in natural populations of Castanopsis fargesii (Fagaceae). Heredity 113, 533–541 (2014). https://doi.org/10.1038/hdy.2014.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.58
This article is cited by
-
Analysis of spatiotemporal variations in the characteristics of soil microbial communities in Castanopsis fargesii forests
Journal of Forestry Research (2020)
-
Ancient east-west divergence, recent admixture, and multiple marginal refugia shape genetic structure of a widespread oak species (Quercus acutissima) in China
Tree Genetics & Genomes (2018)
-
Local adaptation to temperature and precipitation in naturally fragmented populations of Cephalotaxus oliveri, an endangered conifer endemic to China
Scientific Reports (2016)
-
Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites
Scientific Reports (2016)
