
Abstract
The long generation time and large effective size of widespread forest tree species can result in slow evolutionary rate and incomplete lineage sorting, complicating species delimitation. We addressed this issue with the African timber tree genus Milicia that comprises two morphologically similar and often confounded species: M. excelsa, widespread from West to East Africa, and M. regia, endemic to West Africa. We combined information from nuclear microsatellites (nSSRs), nuclear and plastid DNA sequences, and morphological systematics to identify significant evolutionary units and infer their evolutionary and biogeographical history. We detected five geographically coherent genetic clusters using nSSRs and three levels of genetic differentiation. First, one West African cluster matched perfectly with the morphospecies M. regia that formed a monophyletic clade at both DNA sequences. Second, a West African M. excelsa cluster formed a monophyletic group at plastid DNA and was more related to M. regia than to Central African M. excelsa, but shared many haplotypes with the latter at nuclear DNA. Third, three Central African clusters appeared little differentiated and shared most of their haplotypes. Although gene tree paraphyly could suggest a single species in Milicia following the phylogenetic species concept, the existence of mutual haplotypic exclusivity and nonadmixed genetic clusters in the contact area of the two taxa indicate strong reproductive isolation and, thus, two species following the biological species concept. Molecular dating of the first divergence events showed that speciation in Milicia is ancient (Tertiary), indicating that long-living tree taxa exhibiting genetic speciation may remain similar morphologically.
Similar content being viewed by others


Introduction
Angiosperm diversification was termed ‘abominable mystery’ by Charles Darwin because this is likely one of the most complex processes ever investigated by ecologists and biologists. This is because of the difficulty to understand the biogeographical history of specific populations as it depends on interactions between infinite variants of abiotic and biotic phenomena: geological events, climate changes and characteristics, life traits, gene flow between distant populations and sister taxa and so on (Davies et al., 2004). Although most biologists thought that the use of modern tools in phylogenetic taxonomy and molecular ecology in conjunction with data from environmental sciences may provide more accurate insights for inference of biogeographical and evolutionary patterns, the existence of heterogeneous genomic divergence (differentiation across the genome can be highly variable) and the fact that differentiation and speciation are not always synonymous of morphological change (producing cryptic species) can blur the outcomes (Bickford et al., 2006; Nosil et al., 2009).
Especially, addressing speciation in sister taxa is a real challenge complicated by debates on definition of species, as different approaches can reveal different patterns. Most definitions of species are derived from two concepts: (1) the biological species concept (BSC) that defines species as reproductively isolated groups of living organisms (Mayr, 1942), and (2) the phylogenetic species concept (PSC) that groups in a given species individuals forming a monophyletic group (or a clade), according to the monophyly version (De Queiroz, 2007). Because testing for reproductive isolation is difficult and PSC requires many (molecular) traits, historical taxonomy has mostly been based on the morphological similarity of individuals, and ideally the occurrence of multiple diagnostic qualitative traits to define species (Hey, 2006; Mallet, 2010). Nowadays, when multilocus genetic markers are available, large-scale population sampling and Bayesian clustering analyses allow identifying groups of individuals with a shared recent common ancestry, groups that have been reproductively isolated (historically at least) and can be thought as candidate units corresponding to the BSC. Moreover, when DNA sequences are available, they constitute ideal data to identify monophyletic groups of individuals that can be considered as evolutionary units corresponding to the PSC, although incomplete lineage sorting can blur the evidence. Whether historical taxonomic species delimitation converges with the BSC or the PSC has been rarely examined in African tree species (but see Koffi, 2010).
Although different approaches may lead to incongruent results because of reticulated evolution and incomplete lineage sorting, their integration in a pluralistic species concept can help make a consistent decision for taxonomic classification and conservation issues (Freeland et al., 2011). Hence, species delimitation requires to integrate data from molecular phylogeny, classical taxonomy (morphology-based), ecology, historical and contemporaneous biogeography in a way that permits the reconstruction of the evolutionary history of the target taxon (see, for example, Datwyler and Weiblen, 2004; Odee et al., 2012; Scotti-Saintagne et al., 2012). We adopted a pluralistic approach to infer biogeographical history and delimit significant evolutionary units in the African genus Milicia Sim. Taxonomists recognize two sister tree species, M. excelsa (Welw.) C.C. Berg and M. regia (Chev.) C.C. Berg, both called Iroko in the wood trade. Iroko is one of the most important timber woods of Africa. At present, M. regia is viewed as a vulnerable taxon by IUCN (International Union for Conservation of Nature), whereas M. excelsa, a near-threatened species according to the same source, is considered threatened or endangered in some West and East African countries (Ebert, 2004; Ofori and Cobbinah, 2007).
Milicia belongs to the Moreae tribe that also contains Morus, the closest genus to Milicia (Zerega et al., 2005; Nepal, 2012). The most recent common ancestor of Milicia and Morus was estimated between 41 and 72 million years ago (mya) (Zerega et al., 2005). The Moreae have a Eurasian origin and may have primarily diversified during the Palaeocene and the Eocene. At the genus level, few genetic studies have been conducted in Milicia. Using the chloroplast intron trnL and the intergenic spacer trnL-F, Ofori et al. (2001) found no variation in populations collected from the Upper Guinea, the rainforest zone at the western part of the West Africa (Supplementary Figure S1). Ofori et al. (2003) tried to assess genetic differentiation between Milicia species via chloroplast markers, but their populations were assigned to species according to vegetation types because of difficulties in obtaining diagnostic morphological traits of the sampled individuals. Within M. excelsa, Dainou et al. (2010) demonstrated the existence of different gene pools in the Lower Guinea (the rainforest from the Atlantic Ocean until the western side of the Oubangui-Congo River; Supplementary Figure S1), despite extensive gene dispersal (Bizoux et al., 2009), and suggested a relatively recent fragmentation during Pleistocene ice ages. Curiously, Dainou et al. (2010) also showed that West African M. excelsa populations from Benin in the Dahomey gap (the savannah interval separating the Upper Guinean and Lower Guinean forest blocks; Supplementary Figure S1) strongly diverged from Central African samples, despite the fact that M. excelsa presents a continuous range from West to Central Africa. This pattern questions whether M. excelsa individuals from Benin are more closely related to West African M. regia or to Central African M. excelsa populations. In fact, in contrast to the Lower Guinea, we have relatively little information about biogeographical patterns in the Upper Guinea or the Congolia (the humid forest zone eastwards from the Oubangui-Congo River; Supplementary Figure S1; OJ Hardy, unpublished).
The present study aims to disentangle phylogenetic relationships between these sister species using an integrative approach that combines morphological and molecular data. For the present study, we collected samples from most of the distribution range of Milicia in Africa, from Senegal to Tanzania and Democratic Republic of the Congo. Our study represents the largest scale phylogeography study focused on tropical moist forests in Africa. Our investigations were motivated by the following questions: (1) In West Africa, are morphological and genetic markers congruent in delineating species boundary in Milicia? (2) At the scale of Africa, what are the significant evolutionary units within the Milicia species complex and which of them may represent different species? (3) What are the divergence times between evolutionary units and can they be related to the paleodynamics of African rainforests?
Materials and methods
Study taxa
Milicia is a tree up to 35–45 m tall and 1.5 m in diameter. The annual increment in diameter was estimated at 5.7 mm (Durrieu de Madron, 2003), and regular fruiting was observed on trees of at least 60 cm in diameter (Dainou et al., 2012b). M. regia is only distributed in Upper Guinea mostly in the evergreen forests of West Africa, westwards of the Dahomey gap (Supplementary Figure S1). M. excelsa displays a larger geographic distribution from West to East Africa and can occur in a wide range of forest habitats, but is more abundant in semi-deciduous forests (Supplementary Figure S1). The range of the two taxa overlaps from the central part of Liberia to the East of Ghana. M. excelsa and M. regia are morphologically so similar that they are usually confused in the wood trade (ATIBT, 2010) and even by researchers and botanists (Chevalier, 1917; Schnell, 1946; Ofori et al., 2003). Their woods do not present significant differences (White, 1966).
Very little information is available regarding the ecology of M. regia (Dainou et al., 2012a). M. excelsa is a pioneer species with increasing population densities from evergreen forests to semi-deciduous forests (Nichols et al., 1998). Both species are dioecious. M. excelsa is wind pollinated and its seeds are mostly dispersed by birds and the fruit-bat Eidolon helvum (Dainou et al., 2012b). In West Africa, the two species flower and fruit at the same period from January to March (Berg, 1977; Nyong'o et al., 1994).
Sampling and DNA extraction
Samples were obtained from 849 individuals in 17 countries. These included 550 individuals previously studied in Dainou et al. (2010) and sampled from the Lower Guinea and Benin (Dahomey gap) and 46 collected from herbarium collections at the National Botanical Garden of Belgium. During the sampling work, collaborators did not pay attention to the taxonomic identity of individuals. Whenever possible, samplers were asked to collect one entire leaf per individual especially for West African specimens in order to characterize leaf traits, but this was not always possible and many samples consist of a fragment of limb or cambium tissue. The morphological characterization of herbarium vouchers was conducted directly on specimens, as we could not collect entire leaf samples from herbarium collections. Fresh materials were silica-dried. DNA was extracted using the DNeasy Plant Mini kit (QIAGEN, Venlo, The Netherlands).
Genotyping and sequencing
Nuclear microsatellites (nSSRs) were genotyped on the whole sample through seven loci: Mex51, Mex63, Mex81, Mex95, Mex137, Mex163a and Mex202 (Ouinsavi et al., 2006). Amplification conditions were as reported in Bizoux et al. (2009). Genotyping was repeated until each individual is scored for at least six loci.
The amplification and polymorphism of nuclear DNA sequences (nDNA) were tested at 12 gene regions: ADH, GScp, ADHc, RnI1, TPI 6rn/4rn, LF4 Cl1R, LF4 Cl4R, Agt1, Apg1, At103 (Li et al., 2008), and PEPC E1/E2 and PEPC E2/E3 (J Duminil et al., unpublished). Three of them successfully amplified, Agt1, Apg1 and At103, but only the latter was polymorphic and then selected for further analyses. For the plastid genome (pDNA), two intergenic regions, psbA-trnH and trnC-ycf6, were selected. The three nDNA and pDNA sequences were successfully amplified on fresh material but not on the herbarium vouchers in most cases. To overcome this problem, we defined new internal primers that amplified a significant proportion of herbarium samples. The new primer sequences were as follows: 5′-TTCTTGTTCTATCACGAAGTTTGG and 3′-AATCCACTGCCTTGATCCAC (411 bp) for psbA-trnH, 5′-CCGGACGGAGAATCTAATGA and 3′-TCTTTTGTCGCCCTTCATTT (496 bp) for trnC-ycf6 and 5′-CCTGAAACACGATTTGAGAGG and 3′-AGAACTGGTGRCAGGATGAG (342 bp) for At103. PCRs were performed in a thermal cycler PTC 200 (MJ-Research, Waltham, MA, USA). psbA-trnH was amplified in a total volume of 25 μl with the Phusion polymerase (Finnzymes Espoo, Finland) following the manufacturer’s protocol. PCR conditions for trnC-ycf6 were as described in Dainou et al. (2010) and were also applied for At103. From the subsample of 260 individuals selected for sequencing, 195, 225 and 202 were successfully amplified for psbA-trnH, trnC-ycf6 and At103, respectively, with 172 individuals common to the three fragments. In order to test whether Milicia forms a monophyletic group, we also included a sample from Morus indica in the sequencing processes.
Morphological characterization
In an exhaustive literature review of morphological traits that differentiate M. regia and M. excelsa, Dainou et al. (2012a) reported that there could be a slight difference in female flowers according to Aubréville (1959), whereas most botanists agree that ‘in adult specimens the only differences can be found in the leaf venation and the indument’ (Berg, 1977; pg 349). One qualitative and three quantitative leaf characters distinguish the adult specimens of the two species (Hawthorne and Jongkind, 2006). (1) Leaf lower surface is soft to touch (variable Soft) because of the presence of rounded areoles containing microscopic hairs in M. excelsa (Soft=1), whereas this feature is absent in M. regia (Soft=0). (2) The number of pairs of secondary veins (variable N_veins) should not exceed 11 in M. regia, whereas it ranges from 12 to 22 in M. excelsa. However, Berg (1977) and Hawthorne and Jongkind (2006) reported a few cases of M. regia leaves displaying up to 14 veins and M. excelsa leaves with only 10 veins. In such cases, the authors implicitly used only the character Soft to identify each species. (3) The length/width ratio of limb (variable L/w_limb) equals ∼1.4 in M. regia and ∼1.7 in M. excelsa. (4) The mean distance between two contiguous veins (variable D_veins) in M. excelsa is expected to be about half the value found in M. regia specimens (∼1 cm).
We characterized these four traits on a subsample of 114 individuals with entire leaves that came from 11 countries of West and Central Africa. We performed a principal component analysis using STATISTICA 6.0 (StatSoft, 2004) considering the quantitative variables L/w_limb, N_veins and D_veins. The qualitative variable Soft was integrated in the principal component ordination using two different symbols to represent individuals with Soft=0 or 1.
Identification of genetic clusters
Identification of the number of genetic clusters K was based on variation at nSSRs, using the Bayesian clustering algorithm implemented in TESS 2.3.1 (Chen et al., 2007) without prior information of the morphological description. We applied the method as described in Dainou et al. (2010) with some slight differences. Only the admixture model was run with an interaction parameter ψ=0, which means that no spatial information was included in the analysis. For each fixed value of the number of clusters (Kmax) ranging from 2 to 10, we performed 10 runs with a burn-in of 20 000 for a total number of sweeps of 100 000. The best value of K was determined after plotting values of log-likelihood and deviance information criterion against Kmax: for both approaches, the plateau starting point indicated the number of clusters K (Pritchard et al., 2000 and François and Durand, 2010, respectively). We also applied the method in Evanno et al. (2005) to confirm the previous methods. The program CLUMPP (Jakobsson and Rosenberg, 2007) was then used to summarize the five most reliable simulations for the best K, and to estimate averaged cluster membership coefficients q for each individual. At this step, an individual was assigned to a given cluster whenever q>0.5.
Phylogeography and phylogeny at pDNA and nDNA
The following analyses were performed on the subsample of 172 individuals sequenced at all pDNA and nDNA fragments. Two sets of haplotypes were defined based on variation at (1) At103, and (2) over the two plastid sequences psbA-trnH/trnC-ycf6 (Supplementary Tables S1 and S2). Median joining networks were constructed with NETWORK 4.6 (Bandelt et al., 1999).
Bayesian inferences of phylogenetic relationships were performed using BEAST 1.7.3 (Drummond and Rambaut, 2007). M. indica was used as outgroup. First, JMODELTEST 0.1.1 (Posada, 2008) indicated F81 and HKY as the likely substitution models for psbA-trnH / trnC-ycf6 and At103, respectively, after likelihood ratio tests. Then, for each type of genomic sequence, BEAST was run and set up as follows: (1) for the model F81, the appropriate code was imported from BEAST webpage (http://beast.bio.ed.ac.uk/Substitution_model_code) and we modified the input file accordingly; (2) we assumed an uncorrelated lognormal relaxed molecular clock (in general, the uncorrelated relaxed clock models perform well even if the analyzing data are clocklike; Drummond et al., 2006) and a Yule tree prior; (3) the tree root height was constrained with the age of the MRCA of Milicia and Morus (Zerega et al., 2005), assuming a normal distribution with a mean of 56 mya and the s.d. of 9 mya (corresponding to 95% of the distribution lying between 41 and 71 mya); (4) the Markov Chain Monte Carlo was run independently five times for 50 million generations each, sampling 1 tree every 2000 generations. The output files of BEAST were checked with TRACER considering the effective sample size of >100 and combined using LOGCOMBINER, both programs distributed with BEAST. TREEANNOTATOR 1.7.3 (also distributed with BEAST) was then used to select the maximum clade credibility tree (the tree with the highest sum of posterior probabilities on its internal nodes) with the height of nodes calculated as the mean height of all sampled trees for that clade. FIGTREE 1.3. (A Rambaut; http://tree.bio.ed.ac.uk/software/figtree/) was finally used to visualize the phylogenetic trees.
Diversity and differentiation parameters: detecting phylogeographic signal
The genetic clusters determined from nSSRs data were used as basis for computing diversity and differentiation parameters for each type of marker. Relative to the degree of genetic diversity, the following genetic diversity parameters were computed for each genetic cluster: A0, total number of haplotypes (pDNA and nDNA) or alleles (nSSRs); RS, haplotypic (pDNA and nDNA) or allelic richness (nSSRs); %Rpriv, percentage of private haplotypes or alleles; and HT and VT, haplotypic diversity based on unordered and ordered haplotypes, respectively (after Pons and Petit, 1996). Differentiation at nSSRs between pairs of genetic clusters was computed using FST and tested with FSTAT 2.9.3.2 (Goudet, 1995). To check for a phylogeographic signal at nSSRs, we also computed RST that is based on allele size and expected to be larger than FST if mutations (under stepwise model) have contributed to differentiation (Hardy et al., 2003). We also estimated differentiation between clusters and tested for a phylogeographic signal at pDNA and nDNA using GST (for unordered haplotypes) and NST (for ordered haplotypes) (Pons and Petit, 1996). Note that a genetic distance matrix between haplotypes was first inferred following the number of polymorphic characters that differed in state between each pair of haplotypes for pDNA and nDNA. In both cases, 10 000 permutations were performed. Unless specified, computations described above were carried out using SPAGEDI 1.3 (Hardy and Vekemans, 2002) except haplotypic and allelic richness computed using the rarefaction procedure implemented in HP-RARE 1.1 (Kalinowski, 2005). RS and %Rpriv were computed for subsamples of seven genes and 55 individuals at pDNA and nDNA sequences and nSSRs, respectively, taking into account the minimum size of the clusters for each type of markers.
Results
Morphology-based species delimitation
The two first axes of the principal component analysis explained 93.05% of the total variance (Figure 1). The first axis, which was mainly determined by the variable N_veins (45.3% of contribution), split individuals in two groups, one containing samples from Central Africa, Benin and Nigeria presenting soft leaves (Soft=1), and the other one containing samples from Senegal to Sierra Leone and Ivory Coast, without hairs underneath the leaves (Soft=0), although there is a small overlap on the principal component analysis plane. As a consequence, the characters N_veins, D_veins and L/w_limb display significantly different mean values between individuals with Soft=0 and those with Soft=1 but their distributions overlap (Table 1). For example, four trees with Soft=0 presented 14–15 pairs of secondary veins that is unexpected in that group.

Principal component analysis (PCA) projection of Milicia individuals based on three leaf traits: L/w_limb (length/width ratio of limb), N_veins (number of secondary veins) and D_veins (distance between two contiguous veins). Black triangles represent samples with Soft=1, a character expected in M. excelsa, whereas red triangles are samples for which Soft=0, a characteristic of M. regia. Country codes: Be, Benin; Ca, Cameroon; Co, Republic of the Congo; Ga, Gambia; Gb, Gabon; Gh, Ghana; Gu, Guinea Bissau et Guinea-Conakry; IC, Ivory Coast; Li, Liberia; Ni, Nigeria; Se, Senegal; Si, Sierra Leone.
Thereafter, as there are no clear species-specific limits for the three quantitative traits, we will assign M. excelsa to individuals with Soft=1 and M. regia to individuals with Soft=0, as Berg (1977) and Hawthorne and Jongkind (2006) suggested. The character Soft allowed us to identify 335 specimens (135 from West Africa and 200 from Central Africa) because any small piece of leaf was enough for that. All individuals assigned to M. regia were distributed from Senegal to Ghana and never occurred in Central Africa. In West Africa, M. excelsa was found from Guinea-Conakry to Nigeria, making the overlap zone of the two species from Guinea-Conakry to Ghana (Supplementary Figure S1). M. regia mostly occurred in the evergreen forest zone, whereas M. excelsa was mostly present in semi-deciduous forests.
The nSSR genetic clusters and relationships with morphological designation
Log-likelihood and deviance information criterion methods (Pritchard et al., 2000 and François and Durand, 2010, respectively) suggested K=5 as the number of clusters, whereas variation of ΔK (Evanno et al., 2005) suggested two solutions: K=2 and K=5 (Supplementary Figure S2). The case of K=2 clusters may be explained by the existence of two sister species in our data set, as the method of Evanno et al. (2005) is known to detect the uppermost structure in situations of strong differentiation. It performs less well on situations of weak and moderate genetic differentiations (Waples and Gaggiotti, 2006). Therefore, we have to consider K=5 clusters as the true number of clusters (K) in our data set. Cluster K1 grouped together all morphology-based M. regia individuals (Figure 2). All West African M. excelsa individuals were grouped into K2. Clusters K3, K4 and K5 were constituted only of M. excelsa specimens from Central Africa. A proportion of 5.4% of the whole sample was not assigned to any cluster, with q never exceeding 0.50 in these cases. When assessing the degree of purity of each cluster through the proportion of admixed individuals for q>0.8, for example, K1 was the purest (82%), followed by K2 and K3 (70%), K4 (35%) and K5 (22%) (Supplementary Figure S3). Clearly, most individuals assigned to K4 or K5 present a significant portion of their genome also assigned to another of the Central African clusters K3, K4 and K5 (Supplementary Figure S4).

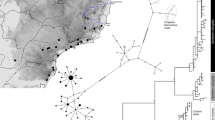
Spatial genetic structure of Milicia populations derived from TESS clustering algorithm for an optimal number K=5 clusters. Black crosses indicate morphology-based M. regia (group of) individuals, whereas the white circles stand for M. excelsa. Codes of the 17 sampled countries, from West to East: Se, Senegal; Ga, Gambia; GBi, Guinea Bissau; GCo, Guinea-Conakry; Si, Sierra Leone; IC, Ivory Coast; Gh, Ghana; Be, Benin; Ni, Nigeria; Ca, Cameroon; Gb, Gabon; Co, Republic of the Congo; CK, Democratic Republic of the Congo; Bu, Burundi; Ta, Tanzania.
One can also assess the hierarchy of clustering when considering the solutions of the clustering algorithm at lower K (Supplementary Figures S4 and S5). For K=2, West African samples (from Senegal to Benin, including all M. regia but also part of M. excelsa) were separated from the rest of the sampling (from Benin to Tanzania). The latter group was further subdivided at K=3 and K=4 (Supplementary Figures S4 and S5), but it was only when K=5 that all M. regia samples form a pure genetic cluster.
Degree of differentiation between the West African M. excelsa cluster K2 and the three clusters from Central Africa (K3 to K5; FST ranged from 0.121 to 0.163, RST from 0.103 to 0.201) were close or in the same range to values found at interspecific level (FST ranged from 0.147 to 0.255, RST from 0.171 to 0.308 between M. excelsa-K1 and any M. excelsa cluster K2 to K5), whereas differentiation was lower among Central African clusters (FST ranged from 0.087 to 0.158, RST from 0.027 to 0.099 between K3, K4 and K5; Table 2). Allele size permutation tests did not detect any phylogeographical signal at nSSR markers (Table 2).
Phylogeographic structure at pDNA and nDNA sequence variation
Whatever genome is considered, M. regia is well differentiated from M. excelsa populations as there is no shared haplotype and, when the trees are rooted, M. regia appears monophyletic (Figures 3a and b). At pDNA fragments, seven haplotypes were restricted to West Africa and, surprisingly, M. excelsa haplotypes in Central Africa belonged to a distinct lineage to the one of West African M. excelsa. When we excluded repetitive sequences (microsatellites) from the analysis, it turned out that only one mutation separates the unique haplotype of M. regia from the most frequent haplotype of M. excelsa in West Africa (Supplementary Figure S6A). Furthermore, at psbA-trnH only, excluding microsatellite sites led to a unique and common haplotype within West African populations, regardless of the taxon (Supplementary Figure S6B). A similar pattern of differentiation was observed at nDNA sequence (M. regia haplotypes forming one clade) except that West and Central African M. excelsa samples shared several haplotypes (Figure 3b). Nevertheless, differentiation indices between the West African cluster M. excelsa-K2 and the Central African clusters M. excelsa-K3 to K5 (GST ranged from 0.479 to 0.716 at pDNA and from 0.201 to 0.319 at nDNA) was nearly as high as between M. excelsa-K2 and M. regia-K1 (GST=0.685 at pDNA and 0.379 at nDNA), and much higher than between Central African clusters (GST ranged from 0.105 to 0.393 at pDNA and from 0.033 to 0.097 at nDNA) (Table 2). There was a significant phylogeographical signal (NST>GST) in the whole data set at both pDNA and nDNA, resulting from the interspecies comparison (Table 2). Within M. excelsa, a significant phylogeographic signal also occurred between the West African cluster K2 and the Central African M. excelsa clusters at pDNA but not at nDNA, whereas no signal of phylogeographical structure was found among Central African populations (Table 2).

Geographical distribution of psbA-trnH / trnC-ycf6 (a) and At103 (b) haplotypes in Milicia and median joining networks. Repetitive sequences were included in these analyses.
When haplotypes at pDNA and nDNA, nSSR-based clusters and morphological assignments (based only on the character Soft) are compared, there is a strong congruence between all types of markers, for the subsample of 172 individuals common to them all (Table 3). All morphology-based M. regia individuals belonged to the lineage formed by pDNA haplotypes H9-H10-H11 and to the cluster K1. Similarly, M. excelsa in West Africa (K2 and far west of K3) was only represented in the lineage formed by pDNA H1-H2-H7-H8. At the cluster level, the pDNA haplotypes H1 and H8 were shared between clusters K2 and K3. However, the three individuals in cluster K3 with these haplotypes were located at the West of Nigeria, closer to Benin than to Cameroon. Therefore at the regional scale, there was no shared haplotype between West Africa and Central Africa for pDNA sequence. In contrast, nDNA sequences displayed shared haplotypes between West and Central Africa.
Variation of genetic diversity among regions and species
West African clusters displayed higher genetic diversity than those of Central Africa at nSSRs, with HT varying from 0.600 to 0.683, whereas it was always <0.490 in Central Africa (Table 2). However, that trend was reversed at nDNA and pDNA sequences. Similarly, haplotypic richness looked higher in Central Africa, but the percentage of private alleles or haplotypes was threefold higher in West African clusters than in Central Africa, with the exception of cluster K5 for pDNA that presented an important proportion of endemic haplotypes (Table 2).
Phylogenetic relationships and timing of divergence
According to both pDNA and nDNA analyses, M. regia is monophyletic. In contrast, M. excelsa appears paraphyletic (Figure 4), at least at pDNA where West African populations of M. excelsa (cluster K2) form a well-supported clade (posterior probability of clade D=0.99; Figure 4a) more closely related to M. regia than to Central African populations of M. excelsa (posterior probability support of the clade B=0.94; Figure 4a). The nDNA phylogenetic tree does not display a M. excelsa clade restricted to West Africa, and although M. excelsa seems paraphyletic in the most likely tree (Figure 4b), there is no strong support (posterior probability of clade J <0.50).

Phylogenetic trees in Milicia based on (a) pDNA (psbA-trnH and trnC-ycf6 intergenic regions) and (b) nDNA (At103 intron) regions. The tree root height was constrained with the age of the MRCA of Milicia and Morus: mean of 56 mya and s.d. of 9 mya (in millions of years ago (mya)) (Zerega et al., 2005). Numbers at the right part of nodes are posterior probabilities (⩽1). The scale bar at the bottom of each figure indicates time period. The estimated mean ages of selected clades are provided in circles along with their 95% highest posterior density (HPD) interval in brackets (empty brackets: no available HPD information as posterior probability was ⩽0.50). Letters A to K are names used in the text for selected nodes.
Molecular dating of the divergence of lineages in the genus Milicia was quite congruent in the two genomes used. The MRCA of Milicia was estimated at 31 and 32 mya (Middle Oligocene) with a wide confidence interval (12–55 mya; clades A and F; Figures 4a and b). Age should correspond to the maximum age of speciation in the genus Milicia. Most of the well-supported major clades within Milicia are dated between 16 and 24 mya (Late Oligocene to Early Miocene; clades B and E in Figure 4a, and clades G and K in Figure 4b). This includes the nDNA well-supported M. regia clade (K) dated at 16 mya. These results should be considered with caution as the confidence intervals were relatively wide because of the fact that we used a single calibration point provided with an important s.d.
Discussion
The present work examined the pattern of genetic differentiation in the African timber tree genus Milicia that is threatened in several African countries because of overexploitation and habitat degradation. Our divergence date estimates showed that Milicia has a Tertiary origin. Despite the tenuous morphological differences between the two species classically recognized by taxonomists, all markers congruently supported the recognition of M. regia (forming one genetic cluster at nSSRs, and monophyletic groups at nDNA and pDNA sequences) that can be distinguished from M. excelsa by the absence of rounded areoles containing microscopic hairs. Differentiation within M. excelsa is more complex because (1) four genetic nSSR clusters were detected: one in West Africa well differentiated from three more related Central African clusters, (2) the West African pDNA clade of M. excelsa is more related to M. regia than to Central African M. excelsa haplotypes, whereas many nDNA haplotypes are shared between West and Central Africa. Hence, West African populations of M. excelsa constitute a questionable group. Hereafter, we first discuss the implications of these results for species delimitation according to the species concepts, and then we question the origin of the differentiation pattern in relation to the history of African vegetation changes.
On phylogenetic relationships and interspecific mating
Under the PSC, a species must be a monophyletic group of organisms. Although DNA sequences show it is the case of M. regia, this does not apply for M. excelsa. The latter could be subdivided in two monophyletic groups according to pDNA but these groups are not supported by nDNA sequences. Hence, under strictly a PSC view, only one species could be recognized in Milicia, with two subspecies or varieties. Whereas phylogenetic analyses support only one species of Milicia, the other findings suggested a more cautious attitude. Three findings tended to confirm an important reproductive barrier between Milicia taxa, a key criterion for the BSC.
First, in West Africa where the two species cooccur, we detected a morphological differentiation for leaf traits. This argument could seem negligible as just leaf traits are used to distinguish the two species (see the next section of the discussion for the importance of using vegetative traits in species delimitation). We think that these slight leaf-trait differences are not insignificant in the case of Milicia for the following reasons. Morphological differentiation was congruent with interspecific genetic divergence. Moreover, assuming that M. regia leaf characters are simply signs of adaptation to evergreen forests would lead to expect such characters in Gabonese evergreen forests too, for example. This genus has likely a Tertiary origin and we should reasonably observe some individuals similar to the morphospecies M. regia in Central African evergreen forests. That was not the case: Central African individuals in our sample, regardless of the vegetation type, had the same M. excelsa leaf traits as individuals from drier forest zones of West Africa (Dahomey gap).
A second argument that rejects the assumption of only one species in Milicia comes from the existence of mutual allelic exclusivity within this genus, in the sense of Doyle (1995) and Flot et al. (2010). This concept states that speciation can be assumed in paraphyletic groups if they have no common allele (mutual exclusivity). Mutual exclusivity is reached in diverging groups generally before reciprocal allelic monophyly, and therefore it should be more suitable than monophyly for species delineation. Under that concept, we should accept two species in Milicia considering the cluster K1 with respect to the others (Figure 5). Rosenberg (2003) showed that the time for a species (for example, M. excelsa) to become reciprocally monophyletic (99% of its loci) is ∼5.3Ne generations (Ne being the population size), assuming that its sister species is already monophyletic at all loci (for example, M. regia). If we assume that Ne≈100 000 individuals (a minimum figure considering the distribution range and population densities; see, for example, Nichols et al., 1998; Fétéké et al., 2004) and a generation time of 100 years in M. excelsa, then it will take ∼53 millions of years before monophyly would be expected at almost all loci. That estimate is much higher than the mean age of maximum divergence time in Milicia, so that the incomplete lineage sorting may be justified. Rejection of reciprocal monophyly for species identification is in line with the unified species concept of De Queiroz (2007) that defines hypothetical species as separately evolving metapopulation lineages, a lineage being a branch and several lineages forming a clade or a monophyletic group. From this point of view, any of the properties derived from other species concepts can be used as a line of evidence to confirm existence of different species, although it is necessary to have several lines of evidence to corroborate the hypothesis of distinct species.

A schematic representation of lineage divergence and speciation that reflects the case of Milicia. The circles with cross inside represent a lineage, whereas the empty circles stand for another one. Different gray colors indicate different haplotypes within each lineage and circles are sized according to their relative frequency. Each Milicia cluster (K1 to K5) is mentioned nearby the divergence phase it reaches with respect to the others. From the starting point to phase 1, the two diverging groups inherit the polymorphism of their ancestor and are distinguishable just by allele frequencies. FST=0.1 is obtained after ∼Ne/5 in the absence of homoplasy, Ne being the effective population size. If effective sizes are enough large, mutation will exert more influence than genetic drift on the time to reach phase 2. At this step, some sequences can reveal a monophyletic group, whereas others may show polyphyly. That is the case of the cluster K2 with respect to K3, K4 and K5. Much later, complete extinctions of lineages may occur leading, for example, to one clear monophyletic group and possibly mutual allelic exclusivity (phase 3): this is observed in K1 with respect to the other genetic clusters. Reciprocal monophyly is reached at a later phase (phase 4), after ∼5Ne generations (Rosenberg, 2003): no pair of Milicia clusters displays such a situation.
Third, interspecific admixture seemed very scarce. Only four individuals presented jointly leaf characters of both species. In addition, using Bayesian approaches for identifying hybrid individuals as implemented in STRUCTURE (Pritchard et al., 2000), TESS (Chen et al., 2007) and NEWHYBRIDS (Anderson and Thompson, 2002), we found only three to six putative hybrids among 46 individuals sampled in the interspecific contact zone, from Guinea-Conakry to Ghana (K Daïnou, unpublished results). However, this low hybrid proportion (6.5–13%) may be biased because the best hybrid zone for Milicia species should be located in the semi-deciduous forest region of Ivory Coast and Ghana that was not sampled enough for the present study. Clearly, additional investigations are required to assess contemporaneous hybridization patterns in Milicia populations.
On the importance of morphological characters for species delimitation
Despite the limited number of distinctive characters in Milicia, it is worth remembering that scarcity of morphological differences does not mean absence of speciation. Of the animal studies, 23% revealed paraphyly or polyphyly, suggesting a much common phenomenon than generally assumed (Funk and Omland, 2003). Investigating monophyly and species boundaries in higher plants is less common because of a more limited number of related studies and fewer attempts to combine morphological and genetic characterizations.
There are very few morphological characters that delimit M. excelsa from M. regia. Past assessment by taxonomists of the existence of two species on the basis of a few vegetative characters may look surprising. The evolution of vegetative characters is often thought to be related to adaptation to the growth environment, so that these characters are viewed as less reliable than floral traits for systematic studies (Ingrouille and Chase, 2004; Das et al., 2007). However, floral traits may be less commonly used than assumed. Grant (1949) observed among 416 genera that although differences in floral traits were employed for classifying 37–40% of bird-, bee- and fly-pollinated plants, this proportion drops up to 4% for wind- and water-pollinated taxa. Plant species with generalist pollination systems are less subject to strong directional selection of floral traits (Johnson and Steiner, 2000), which may explain the absence of floral differentiation in Milicia species. In addition, Berg’s ‘correlation pleiades hypothesis’ has received notable support from various studies (see, for example, Armbruster et al., 1999; Meng et al., 2008). This hypothesis predicted a stronger relationship between floral and vegetative traits in wind-pollinated and generalized flowers than in specialized flowers, because selective pressures affect jointly both categories of characters in the former (covariation is expected as flower components derived from leaves) whereas they are decoupled in the latter (in order to preserve the flower-pollinator fit, regardless of the environment characteristics). Thus, referring exclusively to vegetative traits for species delimitation in Milicia, a wind-pollinated genus, is quite justified.
Phylogeographical structuring: potential paleoenvironment influences
Diversification in gene genealogy can occur in a stable population, independently of environmental perturbations. However, time of coalescence is readily affected by fluctuating population sizes, natural selection and immigration (Freeland et al., 2011).
Zerega et al. (2005) tracked back the biogeographical history and migration routes of most of genera known for Moraceae. They showed that Moraceae may have originated in Eurasia during the Mid-Cretaceous. The two collisions of the landmasses formed by Africa and India with Eurasia at ∼60 and ∼45 mya have created colonization routes for at least four tribes, including Moreae. That period coincided with the first main diversification phase of West African flora, from the Palaeocene to the Mid-Eocene (65–46 mya; Jacobs, 2004; Plana, 2004). All the northern part of the continent including the Sahara was covered by a mosaic of savannahs and forests. This period may be likely the timing of arrival of Milicia ancestor on the continent.
It is interesting to note that the mean ages of the two first divergence events in Milicia took place at the two first major environmental disturbances reported for the continent, after establishment of West African flora at the Mid-Eocene. The start of speciation in Milicia was estimated at∼31–32 mya (mean ages of nodes A and F; Figure 4) that coincided with a sharp cooling period at the Late Eocene to the Early Oligocene (35–31 mya), leading to a major extinction in the hygrophilous African flora (Maley, 1996; Plana, 2004). The second major environmental perturbation occurred from the Early to the Mid-Miocene (23–15 mya): humid vegetation disappeared in the Sahara, the continent moved northwards, down positioning the Equator. The rainforest belt shifted southwards, and the Tethys Sea was closed (Maley, 1996; Jacobs, 2004). The highly supported nodes E, B, G and K in Figure 4 had mean ages included in that range. Obviously, these correlations are just hypotheses as our estimates of divergence times were not accurate enough.
At a regional scale, three clusters were detected in Central Africa. Unlike West Africa, there was no phylogeographical structure at any type of marker in Central Africa. In addition, the degree of differentiation is far lower within Central African clusters comparatively to values noted in West Africa or between West African clusters and those of Central Africa. Dainou et al. (2010) proposed that genetic clusters within Lower Guinea diverged more than 100 000 years ago, possibly during the ice age period lying from 160 000 to 130 000 years ago (Maley, 1996). The spatial organization of M. excelsa clusters in central Africa may result from the past forest fragmentation, possibly at ice ages. Clusters K3 and K4 centered on Cameroon and Gabon, respectively, may be expansions of the Atlantic coastal refuge zones that stretched from the southwestern part of Nigeria to the South of Democratic Republic of the Congo. The North–South divide nearby the Equator between these two clusters has been documented for other forest tree species (Duminil et al., 2010; Debout et al., 2011) and a significant floristic shift has recently been demonstrated at that latitude (Gonmadje, 2012). Cluster K5 centered on the Democratic Republic of the Congo may represent populations spreading from inland forest refuges surrounding the Congo River. The larger extent of K3 and K5 comparatively to the Gabonese cluster K4 may be related to the fact that they are located on more suitable zones for M. excelsa population dynamics (semi-deciduous forests).
In West Africa, making such link between genetic clusters and refuges is possible if we refer to the map in Anhuf et al. (2006) rather than the one proposed by Maley (1996). These authors diverged substantially in mapping the closest forest refuge of the East of the Dahomey gap. According to Maley (1996), a small, well-delimited refuge was established at the southern frontier of Ivory Coast and Ghana, whereas that refuge should have extended up to the extreme eastern boundaries of Ghana, thus partially located in the Dahomey gap. Under the latter scenario, cluster K2 could have extensively been developed eastwards from that refuge. However, phylogeographical signals within West African clusters denoted older isolations than those observed in Central Africa. Then, we should assume that the same places have acted as forest refuges since at least the Late Oligocene-Miocene after tropical forests were established on the continent (Jacobs, 2004; Plana, 2004). There is no proof of such an assumption for that period in West Africa, but elsewhere in East Africa the role of upland areas as refuges many millions years ago was demonstrated (Yemane et al., 1987).
As Milicia populations in West Africa experienced severe exploitation pressures during past centuries, the lower degree of haplotypic richness in this region may be caused by rapid decrease of population density. The higher degree of allelic/haplotypic endemism in West Africa argues for the hypothesis assuming that West African flora may be substantially older than Central African vegetation (suggested in Vande Weghe (2003) and Jacobs (2004)). In the only two other genetic studies of rainforest tree species (Irvingia gabonensis and Terminalia superba) that have included both West and Central African populations, diversity measures also showed that populations from the western African zones exhibit higher genetic diversity than those of the Lower Guinea and the Congolia (Vigneron, 1984; Lowe et al., 2010). A phylogeographical study of Khaya senegalensis, a dry-forest tree species, also revealed that West African groups display higher levels of genetic diversity than those from Sudan and Uganda (Karan et al., 2012).
Conclusions
The present study investigated the pattern of genetic differentiation in Milicia, based on nuclear microsatellites, plastid and nuclear sequences. Five genetic clusters were identified and their origin could be linked to past environmental disturbances. Milicia is a Tertiary genus with two cryptic species exhibiting incomplete lineage sorting. Existence of paraphyly in Milicia can be explained by the time required for isolated species to become reciprocally monophyletic, and this depends on effective population size and generation time that are likely very high in Milicia species. Mutual allelic exclusivity was observed and this tends to confirm the recognition of two species in that genus, despite very scarce morphological differences. Interspecific hybridization looks scarce at present, but this pattern requires further investigations.
Data archiving
The trnC-ycf6 and psbA-trnH plastid DNA sequences have been deposited in GenBank under the accession numbers KF719985–KF720330. The At103 nuclear DNA sequences are available in GenBank under the accession numbers KJ129036–KJ129052. Nuclear microsatellites data have been deposited in DRYAD at http://datadryad.org/resource/doi:10.5061/dryad.77j02.
Accession codes
References
Anderson EC, Thompson EA . (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160: 1217–1229.
Anhuf D, Ledru MP, Behling H, Da Cruz FW, Cordeiro RC, Van der Hammen T et al. (2006). Paleo-environmental change in Amazonian and African rainforest during the LGM. Palaeogeogr Palaeocl 239: 510–527.
Armbruster WS, Di Stilio VS, Tuxill JD, Flores TC, Runk JLV . (1999). Covariance and decoupling of floral and vegetative traits in nine neotropical plants: a re-evaluation of Berg’s correlation-pleiades concepts. Am J Bot 86: 39–55.
ATIBT. (2010). Statistiques 2009. La Lettre de l'ATIBT 32: 4–25.
Aubréville A . (1959) La flore forestière de la Côte d'Ivoire. Deuxième édition révisée—Tome 1. CTFT: Nogent-sur-Marne: Seine (France).
Bandelt H-J, Forster P, Röhl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Berg CC . (1977). Revision of African Moraceae (excl. Dorstenia, Ficus, Musanga, Myrianthus). Bull Jard Bot Natl Belg 47: 267–407.
Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K et al. (2006). Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22: 148–155.
Bizoux JP, Dainou K, Bourland N, Hardy OJ, Heuertz M, Mahy G et al. (2009). Spatial genetic structure in Milicia excelsa (Moraceae) indicates extensive gene dispersal in a low-density wind-pollinated tropical tree. Mol Ecol 18: 4398–4408.
Chen C, Durand E, Forbes F, François O . (2007). Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7: 747–756.
Chevalier A . (1917) Les végétaux utiles d'Afrique tropicale française—La forêt et les bois du Gabon. Challamel: Paris, France.
Dainou K, Bizoux JP, Doucet JL, Mahy G, Hardy OJ, Heuertz M . (2010). Forest refugia revisited: nSSRs and cpDNA sequences support historical isolation in a wide-spread African tree with high colonization capacity, Milicia excelsa (Moraceae). Mol Ecol 19: 4462–4477.
Dainou K, Doucet JL, Sinsin B, Mahy G . (2012a). Identité et écologie des espèces forestières commerciales d'Afrique centrale: le cas de Milicia spp. Biotechnol Agron Soc 16: 229–241.
Dainou K, Laurenty E, Mahy G, Hardy OJ, Brostaux Y, Tagg N et al. (2012b). Phenological patterns in a natural population of a tropical timber tree species, Milicia excelsa (Moraceae): evidence of isolation by time and its interaction with feeding strategies of dispersers. Am J Bot 99: 1453–1463.
Das M, Bhattacharya S, Basak J, Pal A . (2007). Phylogenetic relationships among the bamboo species as revealed by morphological characters and polymorphism analyses. Biol Plantarum 51: 667–672.
Datwyler SL, Weiblen GD . (2004). On the origin of the fig: phylogenetic relationships of Moraceae from ndhF sequences. Am J Bot 91: 767–777.
Davies TJ, Barraclough TG, Chase MW, Soltis PS, Soltis DE, Savolainen V . (2004). Darwin’s abominable mystery: insights from a supertree of the angiosperms. Proc Natl Acad Sci USA 101: 1904–1909.
De Queiroz K . (2007). Species concepts and species delimitation. Syst Biol 56: 879–886.
Debout GD, Doucet JL, Hardy OJ . (2011). Population history and gene dispersal inferred from spatial genetic structure of a Central African timber tree, Distemonanthus benthamianus (Caesalpinioideae). Heredity 106: 88–99.
Doyle JJ . (1995). The irrelevance of allele tree topologies for species delimitation, and a non-topological alternative. Syst Bot 20: 574–588.
Drummond AJ, Ho SY, Phillips MJ, Rambaut A . (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol 4: 699–710.
Drummond AJ, Rambaut A . (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214.
Duminil J, Heuertz M, Doucet JL, Bourland N, Cruaud C, Gavory F et al. (2010). CpDNA-based species identification and phylogeography: application to African tropical tree species. Mol Ecol 19: 5469–5483.
Durrieu de Madron L . (2003). Accroissement diamétrique du bété et de l'iroko. Bois For Trop 275: 83–87.
Ebert SJ . (2004) Silvicultural potential of Milicia excelsa–Working paper No. 18. I-TOO: Freiburg, Germany.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Fétéké R, Nkolong E, Hubert D . (2004) Plan d'aménagement des unités forestières d'aménagement 10.041, 10.042 et 10.044 regroupées. Pallisco: Douala, Cameroon.
Flot JF, Couloux A, Tillier S . (2010). Haplowebs as a graphical tool for delimiting species: a revival of Doyle's ‘field for recombination’ approach and its application to the coral genus Pocillopora in Clipperton. BMC Evol Biol 10: 372.
Freeland JR, Kirk H, Petersen SD . (2011) Molecular Ecology. John Wiley & Sons, Ltd: Oxford, UK.
François O, Durand E . (2010). Spatially explicit bayesian clustering models in population genetics. Mol Ecol Resour 10: 775–784.
Funk DJ, Omland KE . (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with Insights from Animal Mitochondrial DNA. Annu Rev Ecol Evol S 34: 397–423.
Gonmadje CF . (2012) Diversité et biogéographie des forêts d'Afrique centrale atlantique: le cas du massif de Ngovayang (Cameroun). PhD thesis, Université Montpellier 2: Montpellier, France.
Goudet J . (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Grant V . (1949). Pollination systems as isolating mechanisms in Angiosperms. Evolution 3: 82–97.
Hardy OJ, Charbonnel N, Fréville H, Heuertz M . (2003). Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163: 1467–1482.
Hardy OJ, Vekemans X . (2002). Spagedi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hawthorne WD, Jongkind C . (2006) Woody Plants of Western African Forests: A Guide to the Forest Trees, Shrubs and Lianes from Senegal to Ghana. Royal Botanic Gardens: Kew, UK.
Hey J . (2006). On the failure of modern species concepts. Trends Ecol Evol 21: 447–450.
Ingrouille M, Chase MW . (2004). Becoming fruitful and diversifying: DNA sequence phylogenetics and reproductive physiology of land plants. In: Hemsley A, Poole I, (eds) Evolution of Plant Physiology: From Whole Plants to Ecosystems. London Elsevier Academic Press: London, UK. pp 327–342.
Jacobs BF . (2004). Palaeobotanical studies from tropical Africa: relevance to the evolution of forest, woodland and savannah biomes. Philos Trans R Soc B-Biol Sci 359: 1573–1583.
Jakobsson M, Rosenberg NA . (2007). Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Johnson SD, Steiner KE . (2000). Generalization versus specialization in plant pollination systems. Tree 15: 140–143.
Kalinowski ST . (2005). Hp-Rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Karan M, Evans DS, Reilly D, Schulte K, Wright C, Innes D et al. (2012). Rapid microsatellite marker development for African mahogany (Khaya senegalensis, Meliaceae) using next-generation sequencing and assessment of its intra-specific genetic diversity. Mol Ecol Resour 12: 344–353.
Koffi GK . (2010). Etude de la variabilité génétique et de la phylogéographie de Santiria trimera (Burseraceae)-implications pour une conservation durable des forêts humides d’Afrique. PhD thesis, Université Libre de Bruxelles: Brussels, Belgium.
Li M, Wunder J, Bissoli G, Scarponia E, Gazzani S, Barbaro E et al. (2008). Development of COS genes as universally amplifiable markers for phylogenetic reconstructions of closely related plant species. Cladistics 24: 727–745.
Lowe AJ, Harris D, Dormontt E, Dawson IK . (2010). Testing putative African tropical forest refugia using chloroplast and nuclear DNA phylogeography. Trop Plant Biol 3: 50–58.
Maley J . (1996). The African rain forest - main characteristics of changes in vegetation and climate from the Upper Cretaceous to the Quaternary. In: Alexander IJ, Swaine MD, Watling R, (eds) Essays on the Ecology of the Guinea-Congo Rain Forest. The Royal Society of Edinburgh: Edinburgh Vol. 104B, pp 31–73.
Mallet J . (2010). Why was Darwin’s view of species rejected by twentieth century biologists? Biol Philos 25: 497–527.
Mayr E . (1942) Systematics and the Origin of Species from the Viewpoint of a Zoologist. Columbia University Press: New York.
Meng J-L, Zhou X-H, Zhao Z-G, Du G-Z . (2008). Covariance of floral and vegetative traits in four species of Ranunculaceae: a comparison between specialized and generalized pollination systems. J Integr Plant Biol 50: 1161–1170.
Nepal M . (2012). Phylogenetics of Morus (Moraceae) inferred from ITS and trnL-trnF sequence data. Syst Bot 37: 442–450.
Nichols JD, Agurgo FB, Agyeman VK, Wagner MR, Cobbinah JR . (1998). Distribution and abundance of Milicia species in Ghana. Ghana J Forest 6: 1–7.
Nosil P, Funk DJ, Ortiz-Barrientos D . (2009). Divergent selection and heterogeneous genomic divergence. Mol Ecol 18: 375–402.
Nyong'o RN, Cobbinah JR, Appiah-Kwarteng J . (1994). Flowering and fruiting patterns in Milicia excelsa and Milicia regia Welw. Ghana J Forest 1: 19–29.
Odee DW, Telford A, Wilson J, Gaye A, Cavers S . (2012). Plio-Pleistocene history and phylogeography of Acacia senegal in dry woodlands and savannahs of sub-Saharan tropical Africa: evidence of early colonisation and recent range expansion. Heredity 109: 372–382.
Ofori DA, Cobbinah JR . (2007). Integrated approach for conservation and management of genetic resources of Milicia species in West Africa. For Ecol Manage 238: 1–6.
Ofori DA, Swaine M, Leifert C, Cobbinah JR, Price AH . (2001). Population genetic structure of Milicia excelsa characterized by using RAPD and nucleotide sequencing L. Genet Resour Crop Evol 48: 637–647.
Ofori DA, Swaine MD, Cobbinah JR, Price AH . (2003). Genetic diversity and biodiversity conservation guidelines for Milicia species in Ghana. Ghana J Forest 11: 27–38.
Ouinsavi C, Sokpon N, Bousquet J, Newton CH, Khasa DP . (2006). Novel microsatellite DNA markers for the threatened African endemic tree species, Milicia excelsa (Moraceae), and cross-species amplification in Milicia regia. Mol Ecol Notes 6: 480–483.
Plana V . (2004). Mechanisms and tempo of evolution in the African Guineo-Congolian rainforest. Philos Trans R Soc B Biol Sci 359: 1585–1594.
Pons O, Petit RJ . (1996). Measuring and testing genetic differenciation with ordered versus unordered alleles. Genetics 144: 1237–1245.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Pritchard JK, Stephens M, Donnelly PJ . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rosenberg NA . (2003). The shapes of neutral gene genealogies in two species: probabilities of monophyly, paraphyly and polyphyly in a coalescent model. Evolution 57: 1465–1477.
Schnell R . (1946). Sur quelques plantes à usage religieux de la région forestière d'Afrique occidentale. J Société Africanistes 16: 29–38.
Scotti-Saintagne C, Dick CW, Caron H, Vendramin GG, Guichoux E, Buonanici A et al. (2012). Phylogeography of a complex of lowland Neotropical rain forest trees (Carapa, Meliaceae). J Biogeogr 40: 676–692.
StatSoft. (2004) Statistica. StatSoft France: Maisons-Alfort, France.
Vande Weghe JP . (2003) Les forêts d'Afrique Centrale: la Nature et l'Homme. Latoo: Tielt.
Vigneron P . (1984). Polymorphisme enzymatique et variabilité génétique des provenances ivoiriennes et congolaises de Terminalia superba Engl. et Diels. Bois For Trop 204: 41–49.
Waples RS, Gaggiotti O . (2006). What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15: 1419–1439.
White MG . (1966). A comparison of Chlorophora excelsa (Welw.) Benth and Hook (F.) and C. regia A. Chev. (Fam. Moraceae). Commonw Forest Rev 45: 150–153.
Yemane K, Taieb M, Faure H . (1987). Limnogeologic studies on an intertrappean continental deposit from the northern Ethiopian Plateau (37°03′E, 12°25′N). J Afr Earth Sci 6: 91–101.
Zerega NJ, Clement WL, Datwyler SL, Weiblen GD . (2005). Biogeography and divergence times in the mulberry family (Moraceae). Mol Phylogen Evol 37: 402–416.
Acknowledgements
We thank the project PPR 10.000 (Gembloux Agro-Bio Tech; University of Liege), the Fonds Léopold III (asbl, Belgium), and the Fund for Scientific Research of Belgium (FNRS) through grants FRFC no. 2.4.577.10 and FRFC no. 2.4.576.07 for their financial support. We are indebted to David Monticelli, Lambert Y Kouadio, Guillaume Koffi, Bonaventure Sonké, Michel Baudoin, Shango Mutambue, Jean-Pierre Mate, Emilien Dubiez, Pierre Procès, Bertin Kasongo, Pat Stoffelen and Georges Mumbere for their various contributions. We also acknowledge the logging companies Pallisco, SFID, Wijma (Cameroon) and Precious Woods Gabon for facilitating field work, and the National Botanical Garden of Belgium (Meise) for permitting sample collection from herbaria.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Daïnou, K., Mahy, G., Duminil, J. et al. Speciation slowing down in widespread and long-living tree taxa: insights from the tropical timber tree genus Milicia (Moraceae). Heredity 113, 74–85 (2014). https://doi.org/10.1038/hdy.2014.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.5
Keywords
This article is cited by
-
Genetic breaks caused by ancient forest fragmentation: phylogeography of Staudtia kamerunensis (Myristicaceae) reveals distinct clusters in the Congo Basin
Tree Genetics & Genomes (2023)
-
Revisiting the North-South genetic discontinuity in Central African tree populations: the case of the low-density tree species Baillonella toxisperma
Tree Genetics & Genomes (2020)
-
Species delimitation and phylogeography of African tree populations of the genus Parkia (Fabaceae)
Tree Genetics & Genomes (2020)
-
History of the fragmentation of the African rain forest in the Dahomey Gap: insight from the demographic history of Terminalia superba
Heredity (2018)
-
Development of nuclear SNP markers for genetic tracking of Iroko, Milicia excelsa and Milicia regia
Conservation Genetics Resources (2017)
