
Abstract
Communities of organisms inhabiting extreme terrestrial environments provide a unique opportunity to study evolutionary forces that drive population structure and genetic diversity under the combined challenges posed by multiple geogenic stressors. High abundance of an invasive pantropical earthworm (and the absence of indigenous lumbricid species) in the Furnas geothermal field (Sao Miguel Island, Azores) indicates its remarkable tolerance to high soil temperature, exceptionally high carbon dioxide and low oxygen levels, and elevated metal bioavailability, conditions which are lethal for the majority of terrestrial metazoans. Mitochondrial and nuclear markers were used to analyze the relationship between populations living inside and outside the geothermal field. Results showed that Pontoscolex corethrurus (Annelida, Oligochaeta, Glossoscolecidae) to be a genetically heterogeneous complex within the Sao Miguel landscape and is probably differentiated into cryptic species. The population exposed to the hostile soil conditions within the volcanic caldera possesses the lowest within-population mitochondrial diversity but an unexpectedly high degree of nuclear variability with several loci evidencing positive selection, parameters indicative of a genetically unique population only distantly related to conspecifics living outside the caldera. In conclusion, P. corethrurus inhabiting active volcanic soil is a discrete extremophile population that has evolved by tolerating a mixture of non-anthropogenic chemical and physical stressors.
Similar content being viewed by others


Introduction
The study of soil organisms occupying terrestrial geothermal vents provides an unusual opportunity to investigate the dynamics driving population structure and genetic diversity in metazoan communities analogous to those colonizing the better known, but less accessible, deep oceanic hydrothermal vents (McMullin et al., 2007). Geothermal biotopes generically are reducing environments with particular unique features, such as elevated substrate, water and atmospheric elemental composition, together with constant diffuse degassing and high temperatures (Cruz et al., 1999; Viveiros et al., 2009, 2010). Volcanic gases present in the geothermal field under study (Furnas, Azores, Portugal), typically comprise water vapour, carbon dioxide (CO2), hydrogen sulphide (H2S), sulphur dioxide (SO2), hydrogen chloride (HCl), with lesser amounts of hydrogen fluoride (HF) and the radioactive gas radon (Rn) (Silva et al., 2007; Rinaldi et al., 2010). The ephemeral nature of the geothermal field is expected to favour the settlement by species with enterprising colonization abilities (Vrijenhoek, 1997). We have previously collected and identified the native glossoscolecid earthworm Pontoscolex corethrurus (Müller, 1857), from the geothermal field at Furnas. P. corethrurus is a well-known invasive species of most tropical regions, and its global distribution appears to be limited only by temperature (Lavelle et al., 1987). Being a geophagous endogeic species, (Buch et al., 2011) P. corethrurus shows high plasticity in terms of its tolerance to soil physico-chemical characteristics, including moisture and tropical temperature; moreover, it efficiently assimilates low-quality organic matter, thus allowing survival even in very poor soils (Lavelle et al., 1987; Lattaud et al., 1998). The presence and high abundance of this species in the Azorean geothermal field may indicate its relative tolerance to the multifactorial stress challenges inevitably posed by active volcanism. Morphometric analyses of the epidermis of Amynthas gracilis (another invasive earthworm species, albeit a megascolecid, also inhabiting Furnas soil) show that its respiratory exchange surface is ∼50% thinner than the one in conspecifics resident on inactive volcanic soils (Cunha et al., 2011). The complete absence of any lumbricid earthworm species from the ‘undisturbed’ active volcanic soils, in contrast to their ubiquity in a range of different soil types outside the caldera (Cunha, unpublished), gives credence to the notion that tropical invaders are particularly adept at evolving adaptive adjustments.
The extent and landscape-scale distribution of genetic diversity in P. corethrurus populations in the Azores archipelago is neither presently known nor are the history and source(s) of the colonization event(s) known. However, whether the colonization was a singular or multiple events, and whether the main immigration agent was ‘natural’ or anthropogenic, it is probable that the species experienced a genetic bottleneck (Slatkin, 1985, 1987). If this supposition is correct, then it may also be predicted on the basis of the genetic erosion hypothesis (Coors et al., 2009) that the genetic diversity of P. corethrurus within the Furnas volcanic crater is low compared with that of potential source populations living outside the crater. Furthermore, natural selection may have increased genetic divergence between populations inside and outside the crater due to local adaptation to the conspicuously challenging environment instigated by volcanic activity (Slatkin, 1987; Bossart and Prowell, 1998). Sequences derived from the mitochondrial genome (mitochondrial DNA (mtDNA)) and nuclear introns are the most widely used genetic markers both in species delimitation and historical phylogeography. However, discordance between nuclear and mitochondrial gene genealogies is common because of various processes such as differences in mutation rates, the effect of stochastic demographic processes or incomplete lineage sorting (Hebert et al., 2004; Caitlin and John, 2011; Orozco-terWengel et al., 2011). Nonetheless, deep divergence in mtDNA sequences between related individuals is often taken as evidence for the existence of cryptic species (King et al., 2008; Novo et al., 2009; Andre et al., 2010; James et al., 2010). In addition to the mitochondrial markers, methods based on variable PCR amplification such as Amplified Fragment Length Polymorphisms (AFLPs; (Vos et al., 1995)) can provide a rapid and affordable approach to detect polymorphism in a population on a genomic scale (Campbell et al., 2003) and therefore provide a broader genetic characterization and discrimination within and between populations.
The approach in the present study was to characterize and compare the genetic diversity and phylogenetic relationships of three P. corethrurus populations with different exposure histories to active and inactive volcanism on São Miguel Island in the mid-Atlantic Azorean archipelago. Our analyses were based on mtDNA sequence data complemented with 425 genome-wide AFLP markers. The study had two main aims: (i) to determine to what extent the population of invasive earthworms living in stressful active volcanic soil has experienced genetic erosion; and (ii) to seek evidence that the negative effects of genetic erosion (Bijlsma and Loeschcke, 2012) in this relatively small populations are counteracted by gene flow from contiguous populations living under less challenging edaphic conditions.
Materials and methods
Earthworm sources and collection sites
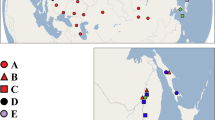
The Azores archipelago comprises nine islands and is located in the North Atlantic Ocean, between 36°45′–39°43′N and 24°45′–31°17′W, at the triple junction of Eurasian, African and North American plates, characterized by a complex tectonic settlement, where seismic and volcanic phenomena are common (Booth et al., 1978). São Miguel is the largest island (757 km2), which presents several active volcanic spots, including fumarolic fields, cold and thermal springs and soil diffuse degassing (Viveiros et al., 2009). The distribution of P. corethrurus in the Azores seems to be restricted to the hot soils of the degassing fields in Furnas and to the enclosed warmth of greenhouses used for pineapple cultivation around the island. Three sampling sites in São Miguel, showing differences in their contemporary geochemical traits, were selected for earthworm capture (Figure 1): (a) Furnas (37°46′24.6″N, 25°18′10.3″W), which displays the most conspicuous degassing and geothermal activity in the entire Azores archipelago; two pineapple greenhouses in (b) Fajã de Baixo (37°45′12.2″N, 25°38′21.3″W), and (c) Vila Franca do Campo (37°45′12.5″N, 25°24′18.3″W). Groups of 20 randomly sampled adults (clitellated) of P. corethrurus were collected by digging and hand sorting during the summer of 2011 at each sampling site. Species anatomy and taxonomical identification was carried out by following the taxonomic key by James (2004). After sampling, the earthworms were immediately transferred to the laboratory, where they were depurated of gut contents by placing them on moistened paper for 36 h. Posterior 2–3 segments (‘tail clips’) were amputated from each individual and preserved in 96% ethanol for later processing.

Sampling sites in São Miguel Island digital elevation model. M.y. million years; T.y., thousand years. A—Sete Cidades volcano, B—Fogo volcano, C—Furnas volcano. Digital elevation model created using the geographical data provided by Secretaria Regional do Ambiente e do Mar (2012).
DNA extraction
DNA was extracted from the caudal tissue samples by phenol–chloroform extraction (Sambrook and Russell, 2001) with the following modifications: the lysis step was accomplished in 2 h using 180 μl of ATL Buffer (Qiagen, Valencia, CA, USA) and 20 μl of Proteinase K (600 mAU ml−1, Qiagen), and the DNA pellet was finally dissolved in 50 μl sterile water. DNA concentration was measured using NanoDrop (ND-1000 spectrophotometer, NanoDrop Technologies, Wilmington DE, USA) to dilute extracts with sterile water to 20 ng μl−1 before PCR. All DNA extracts were stored at −20 °C.
MtDNA cloning
Initially, primers were designed for the amplification of a P. corethrurus cytochrome c oxidase I (COI) fragment based on existing data (AB543229; JN185607). However, using those primers more than one COI sequence per individual was occasionally retrieved. The rationale behind the use of COI and COII is the assumption of character orthology that yield species “barcodes” (Hebert et al., 2003, 2004; Moritz and Cicero, 2004). However, this assumption is violated when paralogous sequences are naively treated as orthologs (for example, nuclear copies of mitochondrial DNA (numts)), leading to incorrect inferences (Funk and Omland, 2003; Song et al., 2008). Further studies are clearly necessary to test this hypothesis (Cunha et al. in preparation). Due to the paucity of mitochondrial sequence information available for P. corethrurus, it was then necessary to retrieve primary sequence information by cloning a large isolated mtDNA fragment (4420 bp) obtained by random size-specific fragmentation of isolated mtDNA. Tissue from one individual was powdered in liquid N2 and used for the extraction of the mitochondrial fraction by sequential centrifugations using a sucrose density gradient modified from Wolstenholme et al. (1972). Briefly, one volume of sample was added to five volumes of isolation buffer composed of 0.3 M sucrose, 1 mm EDTA and 0.01 M Tris–HCl (pH 7.4) maintained at 4 °C with subsequent centrifugation at 480 g for 10 min. The supernatant was removed into a fresh tube and further centrifuged at 9000 g for 12 min. The remaining pellet was incubated with DNAse I (Sigma-Aldrich, Munich, Germany), neutralized with 0.04 M EDTA and centrifuged at 9000 g for 5 min. The final mitochondrial pellet was suspended in 0.15 M NaCl, 0.1 M EDTA and 0.01 M Tris–HCl (pH 8.0) and lysed by adding 0.1 volume of 18% sodium dodecyl sulfate and 0.2 vol of 5 M NaCl. After centrifuging, the supernatant was incubated with RNase A, and total DNA was extracted with phenol–chloroform (Sambrook and Russell, 2001), ethanol precipitated and re-suspended in nuclease-free ultrapure water (Millipore, Bedford, PA, USA). Extracted DNA was purified by column (Qiagen), sheared by nebulization and end repaired according to pJET 2.1 cloning kit (Fermentas, Waltham, MA, USA) recommendations. Efficient random cloning of sheared DNA (∼4.5 kb fragments) was accomplished with the commercial vector pJET 2.1 (Fermentas). Plasmid clone was amplified from both ends with standard primers, digested with HindIII and BglII (Bioline, London, UK) and the resulting fragments cloned as above, and then sequenced with standard pJET kit primers on an ABI PRISM BigDye v3.1 Terminator Sequencing technology (Applied Biosystems, Foster City, CA, USA) on the ABI PRISM 3100 DNA sequencer. Raw sequence traces were confirmed using Finch TV (http://www.geospiza.com/finchTV) before being imported into CLC Main Workbench v6.6 (CLC Bio, Aarhus, Denmark) for the assembling of the full 4420 bp fragment (Genbank accession number: KC665836).
MtDNA genotyping
Two different fragments, including the small ribosomal unit and the NADH dehydrogenase subunit 2 and 3 with some adjacent t-RNAS (Ser, Lys), (see Table 1) were amplified.
For each PCR reaction ∼40 ng DNA template was amplified using 0.4 pmol μl−1 forward and reverse primers, 0.2 mM dNTP mix and 1.25 U μl−1 GO Taq DNA polymerase buffered with 1X Mg-free GO Taq Buffer (Promega UK, Southampton, UK). PCR amplification buffer was supplemented with MgCl2 to achieve a final concentration of 1.75 mM. The reaction was denatured at 95 °C for 3 min, cycled 35 times at 95 °C for 30 s, 30 s at the required primer annealing temperature and 72 °C for 1 min for COI and s-rRNA or 1 min 45 s for the large fragment containing the NAD subunit 2 and NAD subunit 3. This was followed by a 10-min final extension at 72 °C. Before sequencing, PCR clean-ups were performed using Exo-SAP-IT (Amersham Pharmacia, Amersham, UK) reagents. Exonuclease 1 (0.25 μl) and Shrimp Alkaline Phosphatase (0.5 μl) were mixed with the PCR product (10 μl) and incubated at 37 °C for 45 min followed by 80 °C for 15 min. DNA was sequenced using ABI PRISM BigDye v3.1 Terminator Sequencing technology (Applied Biosystems) on an ABI PRISM 3100 DNA automated Sequencer. Raw sequence traces were confirmed using FinchTV (http://www.geospiza.com/finchTV) and aligned in Mega v5.05 (Kumar et al., 2008) for alignment and tree construction.
MtDNA analysis
Phylogenetic relationships between haplotypes were determined by Maximum Likelihood (ML) and Bayesian Inference (BI) using Mega v5.05 (Kumar et al., 2008) and MrBayes v3.2.1, respectively (Huelsenbeck and Ronquist, 2001). Genes were concatenated using DAMBE (Xia and Xie, 2001). JModeltest v0.1.1 (Posada, 2008) was used to select the optimum model of sequence evolution for each individual gene according to the Akaike Information Criterion (Akaike, 1973). Parameters in MrBayes were set to 2 × 106 generations, and 10 000 trees were sampled every 500th generation using the default random tree option to start the analysis under the HKY+G model of sequence evolution inferred with JModeltest. The analysis was performed twice, and all sample points before the plateau phase were discarded as burn-in. The remaining trees were combined to find the maximum phylogenetic tree with the highest posterior probability. An ML tree was constructed using simultaneous NNI to estimate topology. Clade support was evaluated with 500 non-parametric bootstrap replicates (Felsenstein, 1985). A Bayesian analysis of population structure of the mtDNA data was perfomed with the option clustering with linked loci (Corander and Tang, 2007) implemented in BAPS (Corander et al., 2008). BAPS was run three independent times with 10 random values between one and six in the previous upper bound vector. Genetic distances were calculated using p-distance in Mega and median-joining networks were drawn using Network v4.6.1.0 (Bandelt et al., 1999). Arlequin v3.5.1.2 was used to calculate population genetic summary statistcs of the data, that is, fixation indexes, analysis of molecular variance (AMOVA) and a Mantel test between the genetic and geographic distances (Excoffier et al., 2005). The same software was also used to estimate statistics sensitive to demographic changes, that is, Fu’s FS, Tajima’s D and the mismatch distribution. A phylogenetic tree was reconstructed from good quality nucleotide sequences (s-rRNA:455 bp, ND2: 724 bp, ND3: 889 bp) of 60 P. corethrurus (20 per group) earthworms and sequences from Lumbricus terrestris (NC_001673), L. rubellus and Perionyx excavatus (EF494507) as outgroup using Mega.
AFLPs protocol
AFLP analysis was carried out according to Vos et al. (1995) with modifications: two six-base recognition enzymes (PstI and EcoRI) were used, with the aim of reducing the overall number of fragments obtained for each primer combination (Hawthorne, 2001) and increasing the clarity of separation of bands in the fragment analysis output (Mendelson and Simons, 2006). Approximately 50 ng of genomic DNA from each specimen was digested with EcoRI (2.5 U, New England BioLabs, Ipswich, MA, USA (NEB)) and PstI (2.5 U, NEB), Buffer 3 (1 × , NEB) adjusted with water to a final volume of 20 μl and incubated for 2 h at 37 °C. Low-quality DNA samples were excluded and negative controls were run at each step of the genotyping process. Adapters were ligated using 5 pmoles μl−1 of double-stranded EcoRI adapter (5′-CTCGTAGACTGCGTACC-3′ and 5′-AATTGGTACGCAGTC-3′, Sigma-Aldrich), 50 pmoles μl−1 of double-stranded PstI adapter (5′-CTCGTAGACTGCGTACATGCA-3′ and 5′-TGTACGCAGTCTAC-3′, Sigma-Aldrich), ATP (1 mM, Roche Diagnostics, Indianapolis, IN, USA), T4 DNA ligase buffer (1 × , NEB), and T4 DNA ligase (0.7 U, NEB), adjusted to a final volume of 5 μl with water, then added to the double digested genomic DNA (total volume 25 μl) and incubated at 37 °C for 4 h. The digestion–ligation template was diluted (1:10) with low TE buffer (2 M Tris–HCl (pH7.5), 0.5 M EDTA (pH 8)). Pre-selective PCRs contained 2.5 μl diluted template DNA, GoTaq mater mix (Promega), pre-selective EcoRI primer (5′-GACTGCGTACCAATTCA-3′) and PstI primer (5′-GACTGCGTACATGCAGA-3′) (each 2.5 ng μl−1, Sigma-Aldrich), adjusted to a total reaction volume of 20 μl with water. The PCR conditions consisted of an initial denaturation step at 95 °C for 2 min, followed by 30 cycles at 95 °C for 30 s, 56 °C for 30 s and 72 °C for 60 s. The pre-selective template was diluted (1:10) with sterile water for use in the selective amplifications. Selective PCR reactions contained 2.5 μl diluted pre-amplified template DNA, GoTaq mater mix (5 μl, Promega) and EcoRI fluorescent-labelled primer (2.5 ng μl−1) and PstI (15 ng μl−1) primer (Sigma-Aldrich), adjusted to a total reaction volume of 20 μl with water. Four primer combinations were used each with three overhanging nucleotides at the 3′ end (see Table 2). The touchdown thermal cycling programme started with a denaturing step at 95 °C for 2 min, followed by 15 cycles of 95 °C for 30 s, 65 °C* for 30 s (*−0.7 °C each cycle), 72 °C for 60 s followed by 25 cycles of 94 °C for 30 s, 56 °C for 30 s and 72 °C for 60 s. Amplified products were separated on an ABI PRISM 3100 DNA automated Sequencer using GeneScan ROX-500 as size standard (Applied Biosystems).
AFLP scoring and analysis
The fragment size analysis was carried out in Genemarker v2.2.0 (Hulce et al., 2011) using GeneScan 500 size standards (Applied Biosystems) on a panel consisting of all samples. All peaks above 150 r.f.u. (peak intensity identified as a suitable background noise threshold) and between sizes 50–510 bp were scored. Bin positions were manually checked to identify incorrect bin positioning and low quality or noise peaks (irregular shape or pull-ups). Overlapping bin positions were deleted from the data set to avoid ambiguous scoring due to possible size homoplasy of co-migrating fragments (Vekemans et al., 2002). Guidance rules proposed by Crawford et al. (2012) were followed, and individuals with poor quality profiles (failed amplification) were removed from subsequent analysis, as well as fragments that showed a peak above the threshold in the PCR negatives. A total of 60 individuals presented reliable profiles for fragment size scoring. AFLPscore version 1.4b (Whitlock et al., 2008) was used to identify thresholds (relating to average locus peak height and relative peak height across all loci) that resulted in an acceptable mismatch error rate (<5%) while maximizing the number of loci retained for further analysis. Mismatch error rates (Bonin et al., 2007) based on eight repeated genotype profiles (four in Furnas and four in Fajã de Baixo) were calculated using the data filtering option, a locus selection threshold of 500 r.f.u. (13% of the total mean normalized peak height across all loci) and a relative phenotype calling threshold of 150 r.f.u. (10% of the total mean normalized peak intensity across all loci). A binary matrix of retained loci was created in Aflpscore v1.4b for the four primer combinations and a subset were compared with the original electropherograms to check for computational copying errors. Input files for Arlequin v3.5.1.2 (Excoffier et al., 2005) and Structure v2.3.3 (Pritchard et al., 2000) were prepared using AFLPdat (Ehrich, 2006) and putative clone individuals (which only differ by a certain small number of bands) were removed of subsequent analysis. GenAlEx6.41 (Peakall and Smouse, 2006) was used to create a cumulative table of all loci from each individual and transform the data into a binary format and to conduct a principal component analysis (PCA). Additionally, measures of heterozygosity and variation were estimated with GenAlEx v6.41, and a hierarchical AMOVA was carried out in Arlequin v3.5.1.2. Given the uncertainty on the ploidy level of P. corethrurus associated with a possible parthenogenetic reproduction (Muldal, 1952; Jaenike and Selander, 1979), the data was also reanalyzed using an alternative band-based approach (Bonin et al., 2007). As banding patterns of polyploid organisms may not describe an individual’s true genotype (because the presence of a band only reflects the existence of at least one allelic copy), we followed a conservative approach and replaced ‘genotype’ with phenotype as a trait-based description of the organism’s allelic variation (Kosman and Leonard, 2005).
A ploidy independent hierarchical AMOVA of the transformed data under the infinite allele model was carried out using Genodive (Meirmans and Van Tienderen, 2004). We performed a Bayesian analysis of population structure using the software Structure v2.3.3, which can handle dominant data (Falush et al., 2003). Running parameters were left as default under an admixed model with correlated frequencies allowing alpha to be inferred from the data (Falush et al., 2007). Structure was independently run 15 times for values of the number of clusters in the data (K) between 1 and 6 using 1 00 000 steps of burn-in for the MCMC algorithm and 1 00 000 steps of data collection phase. The value of K that best supports the data was inferred with the method of Evanno et al. (2005) calculated on the average of the 15 runs performed for each K value. L(K), the modal choice criterion, was calculated in Structure and the true number of populations (K) was inferred from its maximal value. ΔK, the rate of change in the log probability of data between successive K-values, provides a visual means to easily identify the number of clusters in a sample of individuals (Evanno et al., 2005). The Structure outputs were used to implement the Evanno method with the help of Structure Harvest v0.6.92 (Earl and vonHoldt, 2011). Putative loci under selection were identified using coalescent simulations according to the approach of Beaumont and Nichols (1996), and implemented in the workbench Mcheza (Antao and Beaumont, 2011). The method generates a null distribution of Fst values based on an infinite island model. By comparing for each locus the Fst value against its expected heterozygosity (He), it is possible to detect Øst outlier loci with unusually high Fst. Loci with extreme Fst values are regarded as putative candidates under positive selection, and loci with extremely low Fst values are considered as putative candidates under stabilizing selection. Analyses were performed with the infinite allele mutation model and the significance of the neutral distribution of Fst was tested with 1 00 000 simulations at a significance value P⩽0.05. Additionally, in order to accurately estimate the genetic diversity and population structure, we repeated this analysis using only putative neutral loci.
Results
MtDNA shows deep divergence
Gene descriptive values within the analyzed Pontoscolex population data sets are shown in Table 3. The mitochondrial ND3 and ND2 were the most variable regions showing the highest values of polymorphic sites (189 and 183, respectively) and parsimony informative sites (175 and 167, respectively). The s-rRNA gene was less variable presenting only 26 polymorphic sites (with 26 mutations) and six parsimony informative sites. Furnas presented the lowest haplotype numbers with only one s-rRNA haplotype (out of a total of seven observed haplotypes), four haplotypes for ND3 (out of a total of 11 haplotypes) and two haplotypes for ND2 (out of a total of 13 haplotypes). In general, genetic diversity within the population was conspicuously lower in Furnas earthworms when compared with both Fajã de Baixo and Vila Franca. The multiple sequence alignment of the concatenated sequence data used for phylogenetic analyses was 2068 bp long. It is relevant to note that the use of the s-rRNA gene could not resolve the phylogenetic relationship between the earthworms living inside the geothermal field (Furnas) and the adjacent ‘reference’ populations in pineapple cultures. Only the combined analysis of s-rRNA, ND2 and ND3 presented enough information content to clearly identify the three sampling sites. The high values of ΘW relative to the low values for Θπ may indicate a demographic expansion of the Furnas population (6.76±2.61 and 2.97±1.81, respectively).
The p-distance-based analysis showed that the Furnas population was the most divergent one with an average distance of ∼18% with respect to the out-of-caldera populations, while the latter populations differed from each other by 1% only. Interestingly, uncorrected p-distances of individual genes showed a wide range of variation between the volcanic field and the out-of-caldera populations, with an overall value of 22% for ND3, 28% for ND2 and <1% s-rRNA; these data possibly indicating that the substitution rate in ND2 and ND3 is different from the one presented by s-rRNA. A median-joining network (Bandelt et al., 1999) of the 28 haplotypes found in the concatenated alignment clearly separated the P. corethrurus haplotypes found in Furnas from those found in the two populations living outside the caldera (Figure 2).

Median-joining (MJ) networks showing phylogenetic relationships between populations of P. corethrurus in volcanic and populations living outside the caldera soils. The different colour shades refer to the screened P. corethrurus populations, Furnas haplotypes (dark grey circles), Vila Franca pineapple greenhouse haplotypes (light grey circles), Fajã de Baixo pineapple greenhouse (white circles). MJ network based on the concatenated mitochondrial sequence alignment of 60 individuals. Branch lengths are meaningless. Mutation numbers in italic above branches.
The only exception was one individual from Fajã de Baixo that was more closely related to the haplotypes of Furnas. Consistently with a larger divergence between Furnas and the two populations living outside the caldera populations, the BAPS analysis identified two clusters with a posterior probability of one (Figure 3a). These clusters corresponded to (i) Furnas and the unexpected individual from Fajã de Baixo, and (ii) the remaining individuals of the populations living outside the caldera populations.

The estimated population structure with a summary plot of the estimates of Q based on the mitochondrial data (a) and on the AFLPS data (b). Each individual is represented by a vertical line, which is partitioned into K coloured segments representing the individual’s estimated membership fractions in K clusters (%).
The phylogenetic relationships between the 28 haplotypes in the three Pontoscolex populations as determined with BI are shown in Figure 4. This tree topology was similar to that obtained with ML, except that for a subset of the tree the ML method did not manage to obtain a bootstrap support value >70% (Figure 4). More importantly, the partition between the Furnas haplotypes and the other two sampling sites found by Network was also recovered in this analysis and with both a posterior support of 1 and an ML bootstrap of 100%.

Bayesian phylogenetic hypothesis (BI) based on the three gene fragments amplified for P. corethrurus populations. Furnas haplotypes (dark grey rectangle), Vila Franca haplotypes (light grey rectangle) and Fajã de Baixo haplotypes (white rectangle) are shown. Posterior probability values are shown above the branches and ML bootstrap support values below the branches (unless <70%). The outgroups are placed at the bottom of the figure and are not highlighted.
Both ML and BI trees share common features. Pontoscolex populations sampled in the Fajã de Baixo, Vila Franca and Furnas could be resolved into two distinct genetic lineages. On the one hand, the samples from the ‘greenhouse lineage’ was composed of 97.5% of the individuals sampled from Fajã de Baixo and 100% of those sampled from Vila Franca, on the other hand, the ‘volcanic lineage’ was composed of all the individuals collected from Furnas and the remainder (2.5%) of those sampled from Fajã de Baixo. AMOVA showed highly significant structure (P<0.001) in the data, with most of the genetic variance (91.6%) explained by the differences between volcanic (Furnas) and populations living outside the caldera populations (Fajã de Baixo and Vila Franca; Table 4). Consistent with the inside-caldera versus outside-caldera partitioning of the genetic variance, we found no isolation-by-distance pattern (Mantel test, P=0.66). The mismatch distributions tested for each population and for the two evolutionary clades generated by the phylogenetic analyses (that is, volcanic and populations living outside the caldera) were multimodal and significantly different (P>0.05) from the expected model under a sudden expansion. Additionally, the values of Fu’s FS were not significant for any of the studied populations but, interestingly, Tajima’s D was highly significant and negative for Furnas (−2.39, P=0.001) and Fajã de Baixo (−2.37, P<0.001), suggesting these populations may have expanded demographically in the past or were under the effect of positive selection.
AFLP analysis supports the mtDNA divergence in a shallower timescale
Genetic diversity statistics estimated from AFLP data revealed both within- and among-population differentiation (Table 4). Furnas showed the lowest number of bands (314 bands) when compared with Vila Franca (347 bands) and Fajã de Baixo (404) but presented a higher proportion of private alleles when compared with the populations living outside the caldera populations (Table 5). Vila Franca did not show any private bands and Fajã de Baixo showed only two private bands. Mean expected heterozygosity and Shannon Information Index (I) across populations was 0.167 (s.e.=0.004) and 0.28 (s.e.=0.006), respectively. Among the three populations, the highest value of mean expected heterozygosity (0.197) and I (0.305) were found in Furnas and the lowest in Vila Franca (0.151 and 0.26, respectively) (Table 5). Contrastingly, the larger proportion of polymorphic loci was found in Fajã de Baixo with almost 95% of the total loci, while Furnas presented the lowest proportion of polymorphic loci (74% of the total analyzed loci). The mean percentage of polymorphic loci over all populations was 84%. The highest number of effective alleles was found in Furnas (1.325, s.e.=0.017) and the lowest in Vila Franca (1.204, s.e.=0.01), with an average of 1.246 effective alleles (s.e.=0.007) over all populations. The diversity band-based metrics revealed a similar phenotype number between populations but higher values for the effective number of phenotypes and Shannon index of phenotypic diversity in the Furnas individuals (Supplementary Table S1). The AFLP Bayesian analysis of population structure detected a total of three clusters in the data (Figure 3b; Ln P(D)=−8439.25±7.38, s.d.). The proportion of the total sample genotypes shared by different clusters (that is, each individual’s genotype is represented by a vertical bar in Figure 3b, and the different coloured fragments represent with its colour the cluster to which the genotype belongs) shows that there is a strong evidence of admixture between the observed populations. Cluster 1 mostly consisted of the Furnas population (63%) while on average 75% of the samples from Vila Franca and Fajã de Baixo made cluster 2. Cluster 3 mainly consisted of the remaining individuals from Vila Franca and Fajã de Baixo. Thus, clusters 2 and 3 were dominated by a combination of individuals from both populations living outside the caldera. Interestingly, these results also resemble the outcome of the mtDNA analyses, which also indicated that the two greenhouse reference populations are more similar to each other than either is to the population resident at Furnas.
As for the mtDNA, the AFLP AMOVA analysis showed highly significant differentiation between populations (P<0.001) predominantly as a consequence of deep differences between inside- versus outside-caldera populations. The two reference or greenhouse populations were not significantly different. However, in contrast to the mtDNA findings, the AFLPs showed that most of the genetic variance occurred within populations (79.4%), whereas only 20.6% occurred between populations (Table 4) even when using a ploidy independent model (Supplementary Table S2). The Fst estimate had a value of 0.21 and was highly significant (P<0.001). Consistent with these results, the GenAlex PCA of the individual band pattern clustered most of the individuals from the populations living outside the caldera populations apart from the Furnas individuals, with the first two coordinates explaining 71.7% of the total variation (Figure 5). The PCA plot also showed the Furnas individuals, unlike their greenhouse conspecifics, to be spread rather than tightly clustered, a pattern that reflects the larger genetic variation found in the Furnas population. Correcting the data for unknown allele dosage did not change the PCA patterns (Supplementary Figure S1). Overall, these results indicate separate evolutionary trajectories in Pontoscolex colonizing stressful active volcanic soil compared with counterparts living under relatively unstressed conditions.

The PCA plot of 60 individuals using 425 AFLP markers. Populations are represented by dark grey triangles (Furnas), white rhombi (Fajã de Baixo) and light grey circles (Vila Franca).
Of the 425 loci identified across all samples, 7% correspond to putative loci under selection among which 3% were due to positive selection (Figure 6); (Antao and Beaumont, 2011). We repeated the estimate of genetic diversity and population structure using only putative neutral loci this analysis indicated that removal of the potential outliers had a minimal influence on tree topology and diversity measures (see Supplementary Tables S3, S4 and S5). Interestingly, although the selected loci contributed very little to Furnas population’s genetic diversity, the pairwise Fst comparison between the three populations using only the putative loci under positive selection showed an approximately threefold higher Fst than that using only neutral loci. In fact, ∼57% of the Furnas individuals show a band for the eight putative markers under positive selection, whereas ∼19% of the worms present profiles with variable amounts of bands in them (that is, only 23% of the Furnas worms possess the profile that has a frequency of 95% in worms living outside the volcano).

Mcheza results for all 425 loci across all samples. Most of the loci were found to be putative neutral markers—located in the white region of the figure. The dark grey region above indicates where candidate positive selection loci were found, and the light grey region below indicates where candidate balancing selection loci were found. The putative loci under positive selection are located within the black ellipse in the dark grey area.
Discussion
Mitochondrial-based phylogenetic comparisons showed that haplotypes of the studied populations of P. corethrurus within the volcanic Azorean island are clustered into two major clades. These clades are largely correlated with the contrasting origins of the individual worms (that is, whether living in inactive or active volcanic soils). However, one individual Pontoscolex sampled at the reference site Fajã de Baixo showed to be close to the haplotype typical of worms living in active volcanic Furnas soil, implying that there may be some migration out if not into the caldera mediated either naturally or by anthropogenic means. An alternative hypothesis is that the ‘Furnas-like haplotype’ occurs naturally at low frequency in Pontoscolex populations living outside the caldera soils, and these individual worms could be the founders of the tolerant population inhabiting volcanic soils. Notwithstanding the fact that these two hypotheses require further targeted investigations, the presence of two phylogenetic P. corethrurus lineages is supported by mtDNA AMOVA results. The very high Øst values found for the mtDNA reflect a deep genetic divergence between populations dominated by the differentiation between populations occupying soils differing markedly in stress intensities. Regarding this, the mtDNA and AFLP findings were broadly in agreement. However, although the nuclear DNA showed a large inter-population divergence (Fst∼0.2), it is only a fraction of that observed in the mtDNA (Øst∼0.9). The lower population structure observed with the AFLP data may reflect the relatively recent inadvertent introduction (∼200 years ago) to São Miguel Island from South America in association with tropical crops, such as the pineapple (Stillman and Stillman, 1999). The quantitative discrepancy between the mtDNA- and AFLP-based estimates most likely reflects the expected difference in genealogical lineage sorting between the two data sets due to the fourfold larger Ne size of the nuclear DNA (Orozco-terWengel et al., 2011).
Although the differences between the patterns of variability in the markers studied here may reflect the differences in Ne of the markers and the colonization history of these populations, an explanation behind the low mitochondrial haplotype diversity observed in Furnas remains to be found. The genetic reduction can certainly be part of a small fragmented population’s history, such as those found inhabiting the ‘islands of toxicity’ that typify abandoned metal mine sites (Andre et al., 2010). Endogeic earthworms such as P. corethrurus have low motility and therefore are completely exposed to the surrounding environment. In the case of the volcanic clade in Furnas, this implies having to tolerate the unique combination of hyperthermic, anoxic, hypercarbic, and elevated metal-ion stress, which constitutes a unique extreme environment far different from those soils inside the pineapple greenhouses. Habitat patchiness according to Wiens (1976) is organism-defined and must be considered in terms of the perceptions of the organism rather than those of the investigator. In the case of the lineage from the Furnas geothermal field, it is tempting to interpret the comparatively restricted genetic diversity as a hallmark of stress-driven genetic erosion processes (van Straalen and Timmermans, 2002; Coors et al., 2009) related with natural environmental pressures of the geothermalism having acted upon these populations. Also, the probability that P. corethrurus is a facultative parthenogen (Lavelle et al., 1987) is germane. Parthenogenesis in the inhabitants of the geothermal field would not be surprising, as this reproductive strategy is often associated with critical changes in the environment (Glesener and Tilman, 1978; Lynch and Gabriel, 1983). Parthenogenetic earthworms, most of them known to be polyploid (Muldal, 1952; Jaenike and Selander, 1979), have some fitness advantages that would promote the colonization and the ability to sustain stable populations in conspicuously hostile environments. Higher levels of heterozygosity and exceptionally fit genomes supported by higher reproductive rates (with no clonal population senescence after many generations) as typified by parthenogens such as P. corethrurus enables a quick replacement of losses and make them efficient colonizers when compared with species with obligatory sexual reproduction strategies (Hughes, 1989; Diaz Cosín et al., 2011).
It is noteworthy that gene flow would be unexpected from populations outside the active caldera into the Furnas population owing to the barrier created by the crater, the lower soil temperatures surrounding non-geothermal soils and the extreme conditions inside the geothermal field. Nevertheless, a potential facilitator of anthropologically driven gene flow between these populations of endogeic earthworms could be the pineapple plantations found on the island.
Various mechanisms exist that can drive allelic frequencies in different directions between populations (Nielsen, 2005). One such mechanism is the neutral random process of genetic drift, which can rapidly change allelic frequencies if the effective population size is small (Slatkin, 1985). Another mechanism is positive selection. A selective sweep is an event where an allele that occurs at low frequency in the population becomes adaptive and rises towards fixation due to positive selection. Under this scenario, allelic variants linked to the selected allele also rise in frequency leaving a characteristic genomic signature of a local loss of genetic variation flanked by regions of average heterozygosity (Nielsen, 2005). In our data, eight markers show evidence of being under positive selection. These eight loci are present in the Furnas population while this combination of alleles is extremely rare outside of the volcanic environment (that is, the only individual found outside of the volcano presenting this band profile also carried a Furnas-like mitochondrial haplotype; see haplotype H 5 in Figure 2 and Supplementary Table S6). We hypothesize that the eight AFLP markers occur in relative close proximity to a site that was under positive selection in the recent past in Furnas, which could reveal a case of hitch-hicking (Smith and Haigh, 1974).
The habitat expansion and local adaptation of cosmopolitan animals has been extensively linked to genetic adaptation to counter the biotic and abiotic challenges (Slatkin, 1985). An extensive studied example are the selective sweeps identified in Drosophila populations associated with the X-chromosome (Nurminsky et al., 1995; Harr et al., 2002), in particular the case of hypoxia-tolerant populations (Zhou et al., 2011). The porcine geographic expansion also evidences selective sweeps closely associated with RNA processing and regulation (Groenen et al., 2012).
Some conclusions regarding micro-evolutionary processes can be drawn from the results. The higher intra-lineage diversity of the earthworms living in pineapple greenhouses is indicative of a relatively stationary population that ancestrally presented a high genetic variation or that was founded by multiple introductions (Dupont et al., 2012). Nevertheless, the high number of AFLP markers used in this study compensates for the low number of individuals used and in fact confirms the mitochondrial relationships among the analyzed earthworms.
Most of the Furnas individuals were associated with one cluster irrespective of the marker used, agreeing with the mtDNA evidence that shows a highly divergent and homogeneous P. corethrurus lineage inhabiting the geothermal field, although, in contrast, they showed a higher genetic diversity and also the highest Shannon index value for the AFLP data. The magnitude of these values should be carefully interpreted due to the possible polyploidy of P. corethrurus and therefore cannot be accurately measured. The band-based analysis revealed that between populations the number of phenotypes was found to be very similar, although higher values for phenotypic diversity were always associated with Furnas population (higher effective phenotype number and a higher Shannon index for phenotypic diversity). These results agree with our original analysis based on the assumption of diploidy.
If we consider that this species is polyploid, the consequences of polyploidy for population structure and diversity may be important. For instance, if polyploid migrants move between populations they may reduce genetic divergence beyond the levels expected for diploid organisms, because they carry a larger amount of genetic copies. Additionally, population diversity may be also increase as polyploids can sustain a larger amount of genetic diversity when compared with diploids (Meirmans and Van Tienderen, 2013). Therefore the high diversity found within populations may be indicative of polyploidy and a possible influence for the disparity between the molecular markers used, that is, AFLPs versus mtDNA.
The high variation within population using nuclear markers such as AFLPs and microsatellites is not uncommon in annelids and has been presented even in an exclusively diploid earthworm, such as the nightcrawler Lumbricus terrestris (Richter, 2009; Gailing et al., 2012). Interestingly, the lower diversity of mtDNA found in Furnas individuals could mean a bias restricting mtDNA diversification. On the one hand, it is plausible that the erosive effect of selection on genomic diversity will intensify in populations successfully inhabiting intensely stressful environmental conditions, such as actively volcanic soils. In contrast, an intriguing alternative scenario may pertain where chemical contaminants increase genetic diversity by causing somatic and even genomic mutations (Somers et al., 2002; Hirano and Tamae, 2010), which could explain why Furnas show the highest number of private bands. This finding correlates also with the highest mutations found in mitochondrial data.
The presence of two highly divergent lineages within P. corethrurus in Sao Miguel brings to light the debate of cryptic speciation. Despite its use in ecological studies, this possibility has never been considered in P. corethrurus, and it could have important implications for ecology, conservation and ecotoxicology approaches (King et al., 2008). Speciation is not always necessarily paired with morphological differentiation (Hebert et al., 2004; Ahrens et al., 2007), and cryptic species are typically found in taxa that occur in complex and patchy terrestrial and aquatic habitats (Hebert et al., 2004; Hilário et al., 2010; Boissin et al., 2011; Avrani et al., 2012). In fact, the more studies are performed on annelids the more clear it becomes that cryptic speciation is not an uncommon phenomenon in this phylum (Pfenninger and Schwenk, 2007; Hilário et al., 2010). Earthworms have been the target of several comprehensive phylogenetic studies using both mitochondrial and nuclear markers revealing high intra-species genetic diversity (Velavan et al., 2007; Fernández et al., 2011) and deeply divergent genetic lineages, possibly in some cases corresponding to cryptic species (Novo et al., 2009; Pérez-Losada et al., 2009; Andre et al., 2010). Whether or not these lineages found in P. corethrurus warrant the status of (cryptic) species must wait for further genetic and breeding evidence as well as a more intensive sampling which goes beyond the aims of the present study.
Finally, we recommend that future research on microevolution or macroevolution, biogeography, ecology, conservation and ecotoxicology on these taxa should consider their specific taxonomic status when formulating experimental designs.
Data archiving
Sequence data have been submitted to GenBank: accession numbers KC665735–KC665915. AFLP Genotype data have been submitted to Dryad:doi:10.5061/dryad.fd6jb. Additional results can be found in Supplementary Material.
Accession codes
References
Ahrens D, Monaghan MT, Vogler AP . (2007). DNA-based taxonomy for associating adults and larvae in multi-species assemblages of chafers (Coleoptera: Scarabaeidae). Mol Phylogenet Evol 44: 436–449.
Akaike H . (1973). Information theory as an extension of the maximum likelihood principle. In: Petrov BN, Csaki F (eds) Second International Symposium on Information Theory. Akademiai Kiado: Budapest. pp 267–281.
Andre J, King RA, Stürzenbaum SR, Kille P, Hodson ME, Morgan AJ . (2010). Molecular genetic differentiation in earthworms inhabiting a heterogeneous Pb-polluted landscape. Environ Pollut 158: 883–890.
Antao T, Beaumont MA . (2011). Mcheza: a workbench to detect selection using dominant markers. Bioinformatics 27: 1717–1718.
Avrani S, Ben-Shlomo R, Inbar M . (2012). Genetic structure of a galling aphid Slavum wertheimae and its host tree Pistacia atlantica across an Irano-Turanian distribution: from fragmentation to speciation? Tree Genet Genom 8: 811–820.
Bandelt HJ, Forster P, Röhl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Beaumont MA, Nichols RA . (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond Ser B 263: 1619–1626.
Bijlsma R, Loeschcke V . (2012). Genetic erosion impedes adaptive responses to stressful environments. Evol Appl 5: 117.
Boissin E, Stöhr S, Chenuil A . (2011). Did vicariance and adaptation drive cryptic speciation and evolution of brooding in Ophioderma longicauda (Echinodermata: Ophiuroidea), a common Atlanto-Mediterranean ophiuroid? Mol Ecol 20: 4737–4755.
Bonin A, Ehrich D, Manel S . (2007). Statistical analysis of amplified fragment length polymorphism data: a toolbox for molecular ecologists and evolutionists. Mol Ecol 16: 3737–3758.
Booth B, Croasdale R, Walker G . (1978). A quantitative study of five thousand years of volcanism on Sao Miguel, Azores. Proc R Soc Lond Ser A 288: 271–319.
Bossart J, Prowell PD . (1998). Genetic estimates of population structure and gene flow: limitations, lessons and new directions. Trends Ecol Evol 13: 202–206.
Buch AC, Brown GG, Niva CC, Sautter KD, Lourençato LF . (2011). Life cycle of Pontoscolex corethrurus (Müller, 1857) in tropical artificial soil. Pedobiologia 54: S19–S25.
Caitlin FRM, John W . (2011). What are the consequences of combining nuclear and mitochondrial data for phylogenetic analysis? Lessons from Plethodon salamanders and 13 other vertebrate clades. BMC Evol Biol 11: p300.
Campbell D, Duchesne P, Bernatchez L . (2003). AFLP utility for population assignment studies: analytical investigation and empirical comparison with microsatellites. Mol Ecol 12: 1979–1991.
Coors A, Vanoverbeke J, De Bie T, De Meester L . (2009). Land use, genetic diversity and toxicant tolerance in natural populations of Daphnia magna. Aquat Toxicol 95: 71–79.
Corander J, Marttinen P, Sirén J, Tang J . (2008). Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics 9: 539.
Corander J, Tang J . (2007). Bayesian analysis of population structure based on linked molecular information. Math Biosci 205: 19–31.
Crawford LA, Koscinski D, Keyghobadi N . (2012). A call for more transparent reporting of error rates: the quality of AFLP data in ecological and evolutionary research. Mol Ecol 21: 5911–5917.
Cruz JV, Coutinho RM, Carvalho MR, Oskarsson N, Gislason SR . (1999). Chemistry of waters from Furnas volcano, São Miguel, Azores: fluxes of volcanic carbon dioxide and leached material. J Volcanol Geotherm Res 92: 151–167.
Cunha L, Campos I, Montiel R, Rodrigues A, Morgan AJ . (2011). Morphometry of the epidermis of an invasive megascoelecid earthworm (Amynthas gracilis, Kinberg 1867) inhabiting actively volcanic soils in the Azores archipelago. Ecotoxicol Environ Saf 74: 25–32.
Diaz Cosín DJ, Novo M, Fernández R . (2011). Reproduction of Earthworms: Sexual Selection and Parthenogenesis. In: A. Karaca (Ed) Biology of Earthworms, Springer-Verlag: Berlin Heidelberg. Vol 24, pp 69-86.
Dupont L, Decaëns T, Lapied E, Chassany V, Marichal R, Dubs F et al. (2012). Genetic signature of accidental transfer of the peregrine earthworm Pontoscolex corethrurus (Clitellata, Glossoscolecidae) in French Guiana. Eur J Soil Biol 53: 70–75.
Earl DA, vonHoldt BM . (2011). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
Ehrich D . (2006). AFLPdat: a collection of R functions for convenient handling of AFLP data. Mol Ecol Notes 6: 603–604.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47.
Falush D, Stephens M, Pritchard JK . (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567.
Falush D, Stephens M, Pritchard JK . (2007). Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578.
Felsenstein J . (1985). Phylogenies and the comparative method. Am Nat 125: 1–15.
Fernández R, Almodóvar A, Novo M, Gutiérrez M, Díaz Cosín DJ . (2011). A vagrant clone in a peregrine species: phylogeography, high clonal diversity and geographical distribution in the earthworm Aporrectodea trapezoides (Dugès, 1828). Soil Biol Biochem 43: 2085–2093.
Funk DJ, Omland KE . (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst 34: 397–423.
Gailing O, Hickey E, Lilleskov E, Szlavecz K, Richter K, Potthoff M . (2012). Genetic comparisons between North American and European populations of Lumbricus terrestris L. Biochem Syst Ecol 45: 23–30.
Glesener RR, Tilman D . (1978). Sexuality and the components of environmental uncertainty: clues from geographic parthenogenesis in terrestrial animals. Am Nat 112: 659–673.
Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF et al. (2012). Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491: 393–398.
Harr B, Kauer M, Schlötterer C . (2002). Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc Natl Acad Sci 99: 12949–12954.
Hawthorne DJ . (2001). AFLP-based genetic linkage map of the Colorado potato beetle Leptinotarsa decemlineata: sex chromosomes and a pyrethroid-resistance candidate gene. Genetics 158: 695.
Hebert PDN, Ratnasingham S, de Waard JR . (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B Biol Sci 270 ((Suppl 1)): S96.
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W . (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA 101: 14812.
Hilário A, Johnson S, Cunha M, Vrijenhoek R . (2010). High diversity of frenulates (Polychaeta: Siboglinidae) in the Gulf of Cadiz mud volcanoes: a DNA taxonomy analysis. Deep Sea Res Part I 57: 143–150.
Hirano T, Tamae K . (2010). Heavy metal-induced oxidative DNA damage in earthworms: a review. Appl Environ Soil Sci 2010: 1–7.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Hughes RN . (1989) Functional biology of clonal animals. Chapman and Hall: London, UK.
Hulce D, Li X, Snyder-Leiby T, Liu CSJ . (2011). GeneMarker genotyping software: tools to increase the statistical power of DNA fragment analysis. J Biomol Tech 22 ((Supplement)): S35.
Jaenike J, Selander RK . (1979). Evolution and ecology of parthenogenesis in earthworms. Am Zool 19: 729–737.
James SW . (2004). An illustrated key to the earthworms of the Samoan Archipelago (Oligochaeta: Glossoscolecidae, Megascolecidae, Moniligastridae). Micronesica 37: 1–13.
James SW, Porco D, Decaëns T, Richard B, Rougerie R, Erséus C . (2010). DNA barcoding reveals cryptic diversity in Lumbricus terrestris L., 1758 (Clitellata): resurrection of L. herculeus (Savigny, 1826). PLoS One 5: e15629.
King RA, Tibble AMYL, Symondson WOC . (2008). Opening a can of worms: unprecedented sympatric cryptic diversity within British lumbricid earthworms. Mol Ecol 17: 4684–4698.
Kosman E, Leonard KJ . (2005). Similarity coefficients for molecular markers in studies of genetic relationships between individuals for haploid, diploid, and polyploid species. Mol Ecol 14: 415–424.
Kumar S, Nei M, Dudley J, Tamura K . (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9: 299–306.
Lattaud C, Locati S, Mora P, Rouland C, Lavelle P . (1998). The diversity of digestive systems in tropical geophagous earthworms. Appl Soil Ecol 9: 189–195.
Lavelle P, Barois I, Cruz I, Fragoso C, Hernandez A, Pineda A et al. (1987). Adaptive strategies of Pontoscolex corethrurus (Glossoscolecidae, Oligochaeta), a peregrine geophagous earthworm of the humid tropics. Biol Fertil Soils 5: 188–194.
Lynch M, Gabriel W . (1983). Phenotypic evolution and parthenogenesis. Am Nat 122: 745–764.
McMullin ER, Bergquist DC, Fisher CR . (2007). Metazoans in extreme environments: adaptations of hydrothermal vent and hydrocarbon seep fauna. Gravitat Space Biol J 13: 13–23.
Meirmans PG, Van Tienderen PH . (2004). GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Meirmans PG, Van Tienderen PH . (2013). The effects of inheritance in tetraploids on genetic diversity and population divergence. Heredity 110: 131–137.
Mendelson TC, Simons JN . (2006). AFLPs resolve cytonuclear discordance and increase resolution among barcheek darters (Percidae: Etheostoma: Catonotus). Mol Phylogenet Evol 41: 445–453.
Moritz C, Cicero C . (2004). DNA barcoding: promise and pitfalls. PLoS Biol 2: e354.
Muldal S . (1952). The chromosomes of the earthworms: I. The evolution of polyploidy. Heredity 6: 56–76.
Müller F . (1857). Description of a new earthworm (Lumbricus corethrurus). Ann Mag Nat Hist 20: 13–15.
Nielsen R . (2005). Molecular signatures of natural selection. Annu Rev Genet 39: 197–218.
Novo M, Almodóvar A, Díaz-Cosín DJ . (2009). High genetic divergence of hormogastrid earthworms (Annelida, Oligochaeta) in the central Iberian Peninsula: evolutionary and demographic implications. Zool Scr 38: 537–552.
Nurminsky DI, Nurminskaya MV, De Aguiar D, Hartl DL . (1995). Selective sweep of a newly evolved sperm-specific gene in Drosophila. Nature 396: 572–575.
Orozco-terWengel P, Corander J, Schlotterer C . (2011). Genealogical lineage sorting leads to significant, but incorrect Bayesian multilocus inference of population structure. Mol Ecol 20: 1108–1121.
Peakall R, Smouse PE . (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295.
Pérez-Losada M, Ricoy M, Marshall JC, Domínguez J . (2009). Phylogenetic assessment of the earthworm Aporrectodea caliginosa species complex (Oligochaeta: Lumbricidae) based on mitochondrial and nuclear DNA sequences. Mol Phylogenet Evol 52: 293–302.
Pfenninger M, Schwenk K . (2007). Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evol Biol 7: 121.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945.
Richter K . (2009) Genetic Structure in European Populations of the Earthworm Lumbricus terrestris. Kassel University Press: Kassel, Germany.
Rinaldi AP, Viveiros F, Vandemeulebrouck J, Todesco M . (2010) Effect of atmospheric conditions on soil diffuse degassing. Geophysical Research Abstracts EGU General Assembly: Vienna, Austria vol. 12, pp 2204.
Sambrook J, Russell DW . (2001) Rapid isolation of yeast DNA. In: Sambrook J, Russel DW, (eds) Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory: New York, NY, USA. pp 631–632.
Silva C, Ferreira T, Viveiros F . (2007) Radon (222Rn) soil gas measurements at Furnas Volcano (S. Miguel Island, Azores). Geophysical Research Abstracts EGU General Assembly: Vienna, Austria vol. 9, pp 08372.
Slatkin M . (1985). Gene flow in natural populations. Annu Rev Ecol Syst 16: 393–430.
Slatkin M . (1987). Gene flow and the geographic structure of natural populations. Science 236: 787.
Smith JM, Haigh J . (1974). The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35.
Somers CM, Yauk CL, White PA, Parfett CLJ, Quinn JS . (2002). Air pollution induces heritable DNA mutations. Proc Natl Acad Sci 99: 15904–15907.
Song H, Buhay JE, Whiting MF, Crandall KA . (2008). Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci 105: 13486.
Stillman YK, Stillman NA . (1999) From Iberia to diaspora: studies in Sephardic history and culture. In: David SK (Ed) Brill's Series in Jewish Studies, Brill Academic: Leiden. vol 19, pp 1-547.
van Straalen NM, Timmermans MJTN . (2002). Genetic variation in toxicant-stressed populations: an evaluation of the “genetic erosion” hypothesis. Hum Ecol Risk Assess 8: 983–1002.
Vekemans X, Beauwens T, Lemaire M, Roldán-Ruiz I . (2002). Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol Ecol 11: 139–151.
Velavan T, Schulenburg H, Michiels NK . (2007). Development and characterization of novel microsatellite markers for the common earthworm (Lumbricus terrestris L.). Mol Ecol Notes 7: 1060–1062.
Viveiros F, Ferreira T, Silva C, Gaspar JL . (2009). Meteorological factors controlling soil gases and indoor CO2 concentration: a permanent risk in degassing areas. Sci Total Environ 407: 1362–1372.
Viveiros F, Cardellini C, Ferreira T, Caliro S, Chiodini G, Silva C . (2010). Soil CO2 emissions at Furnas volcano, São Miguel Island, Azores archipelago: Volcano monitoring perspectives, geomorphologic studies, and land use planning application. J Geophys Res 115: B12208.
Vos P, Hogers R, Bleeker M, Reijans M, Lee T, Hornes M et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407.
Vrijenhoek R . (1997). Gene flow and genetic diversity in naturally fragmented metapopulations of deep-sea hydrothermal vent animals. J Hered 88: 285–293.
Whitlock R, Hipperson H, Mannarelli M, Butlin R, Burke T . (2008). An objective, rapid and reproducible method for scoring AFLP peak-height data that minimizes genotyping error. Mol Ecol Res 8: 725–735.
Wiens JA . (1976). Population responses to patchy environments. Annu Rev Ecol Syst 7: 81–120.
Wolstenholme DR, Kirschner RG, Gross NJ . (1972). heat denaturation studies of rat liver mitochondrial DNA: a denaturation map and changes in molecular configurations. J Cell Biol 53: 393–406.
Xia X, Xie Z . (2001). DAMBE: software package for data analysis in molecular biology and evolution. J Hered 92: 371–373.
Zhou D, Udpa N, Gersten M, Visk DW, Bashir A, Xue J et al. (2011). Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc Natl Acad Sci 108: 2349–2354.
Acknowledgements
We thank Paulo Melo and Ana Valentim for the help during the field work, José Talavera for the taxonomical identification of the studied specimens and Joanna Bluemel and Edward Brede for the technical details on AFLP analysis. This study was financially supported by CIRN (University of the Azores), DRCT (Government of the Azores) and by the FCT project PTDC/AAC-AMB/115713/2009. LC was supported by a Doctoral grant from DRCT (M312/F/029/2007). MN was supported by a postdoctoral fellowship from the Spanish Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Cunha, L., Montiel, R., Novo, M. et al. Living on a volcano’s edge: genetic isolation of an extremophile terrestrial metazoan. Heredity 112, 132–142 (2014). https://doi.org/10.1038/hdy.2013.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.84
Keywords
This article is cited by
-
Genetic evidence of multiple introductions and mixed reproductive strategy in the peregrine earthworm Pontoscolex corethrurus
Biological Invasions (2020)
-
Aporrectodea caliginosa, a relevant earthworm species for a posteriori pesticide risk assessment: current knowledge and recommendations for culture and experimental design
Environmental Science and Pollution Research (2018)
-
Phylogeography of Eisenia nordenskioldi nordenskioldi (Lumbricidae, Oligochaeta) from the north of Asia
Polar Biology (2018)
