
Abstract
Although homoploid hybrid speciation in plants is probably more common than previously realized, there are few well-documented cases of homoploid hybrid origin in conifers. We examined genetic divergence between two currently widespread pines in Northeast China, Pinus sylvestris var. mongolica and Pinus densiflora, and also whether two narrowly distributed pines in the same region, Pinus funebris and Pinus takahasii, might have originated from the two widespread species by homoploid hybrid speciation. Our results, based on population genetic analysis of chloroplast (cp), mitochondrial (mt) DNA, and nuclear gene sequence variation, showed that the two widespread species were divergent for both cp- and mtDNA variation, and also for haplotype variation at two of eight nuclear gene loci surveyed. Our analysis further indicated that P. sylvestris var. mongolica and P. densiflora remained allopatric during the most severe Quaternary glacial period that occurred in Northeast China, but subsequently exhibited rapid range expansions. P. funebris and P. takahasii, were found to contain a mixture of chlorotypes and nuclear haplotypes that distinguish P. sylvestris var. mongolica and P. densiflora, in support of the hypothesis that they possibly originated via homoploid hybrid speciation following secondary contact and hybridization between P. sylvestris var. mongolica and P. densiflora.
Similar content being viewed by others


Introduction
Hybridization is widespread in plants and has an important role in plant diversification and evolution (Abbott, 1992; Rieseberg et al., 2003; Arnold, 2006). Hybridization occurs in situations of sympatry and parapatry, often following range shifts and secondary contact between closely related and formerly allopatric species (Song et al., 2002, 2003; Wachowiak and Prus-Głowacki, 2008). Hybridization can result in introgression of alleles across species boundaries and sometimes the origin of a new introgressant lineage (Currat et al., 2008; Kim et al., 2008). Additionally, it can trigger the origin of a new hybrid species through allopolyploid or homoploid hybrid speciation (Rieseberg, 1997; Mallet, 2007; Abbott et al., 2010). Whereas allopolyploid speciation is common in plants, homoploid hybrid speciation is regarded as rare (Rieseberg, 1997; Gross and Rieseberg, 2005), although more examples of homoploid hybrid species are likely to be discovered in the future now that genetic resources are readily available for testing their occurrence.
In contrast to allopolyploid speciation, which is relatively easy to identify from chromosomal and molecular data, homoploid hybrid speciation is difficult to detect unambiguously (Rieseberg, 1997; Mallet, 2007). Nevertheless, a small but increasing number of cases has been documented recently across a range of plant, animal and fungal taxa (Gross and Rieseberg, 2005; Mallet, 2007; Abbott et al., 2010). It is well known that many closely related pine species hybridize, producing fertile and vigorous hybrids that combine morphological traits and/or genetic components of both parental species (Mirov, 1967; Wachowiak and Prus-Głowacki, 2008). Although several Asian pine species are thought to be of hybrid origin (Mirov, 1967), only Pinus densata has been rigorously analyzed and shown to be the homoploid hybrid of Pinus tabulaeformis and Pinus yunnanensis (Wang et al., 1990; Song et al., 2002, 2003, 2011; Ma et al., 2006).
Here, we investigate mitochondrial (mt), chloroplast (cp) and nuclear DNA variation at the population level within four closely related pine species in Northeast China that have the same chromosome number (2n=24; Zhang and Li, 1984). Two of these species, Pinus sylvestris var. mongolica and Pinus densiflora, are dominant components of natural coniferous forests in Northeast China (Wu, 1995), a region considered not to have been glaciated during the Quaternary (Hewitt, 2000). However, climatic oscillations during the Quaternary may have caused these indigenous coniferous forests to retreat to refugia during glacial periods and undergo range expansions during interglacials (Wu, 1995; Aizawa et al., 2007; Chen et al., 2008) as recorded for forest trees in other regions (for example, Petit et al., 2003; Naydenov et al., 2007). Such range shifts can stimulate allopatric divergence during periods of range fragmentation across different glacial refugia (Hewitt, 2004), but provide opportunities for interspecific hybridization following contact during interglacials, although the possibility of subsequent homoploid hybrid speciation has rarely been examined or reported (Abbott and Brochmann, 2003). P. sylvestris occurs widely from Europe to Eastern Asia with var. mongolica being widely distributed in mountainous areas of the northern part of Northeastern China. In contrast, P. densiflora occurs only in Northeast China, Korea and Japan, being found more to the south in Northeastern China and in the Shandong Peninsula (Cheng and Fu, 1978) (Figure 1). A phylogenetic analysis based on four cpDNA markers, previously indicated that P. sylvestris and P. densiflora comprise a pair of sister species within a small pine monophyletic lineage (Wang et al., 1999). They are distinguished by several morphological traits, including differences in needle tips and number of resin canals (Table 1). Phylogeographic analysis of P. sylvestris suggests that the East Asian populations became established following postglacial range expansion of the species from glacial refugia in central Asia (Naydenov et al., 2007).

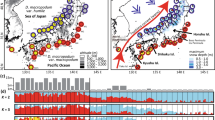
Distributions and networks of chlorotypes (a, c) and mitotypes (b, d) among four pine species in Northeast China. The color of the circumference of circles in (a) and (c) indicates each species and in (b) and (d) is proportional to the frequency of each chlorotype or mitotype in each species. The color filled in circles in (a) and (c) is proportional to the frequency of each chlorotype or mitotype in each population.
The two other pine species investigated here, Pinus funebris and Pinus takahasii, have very narrow distributions and might potentially have originated by homoploid hybrid speciation following hybridization between P. sylvestris var. mongolica and P. densiflora (Cheng and Fu, 1978). P. funebris occurs in a restricted region of the eastern Changbai Mountains where it grows at elevations between 800 and 1600 m, partly overlapping with P. densiflora, which occurs below 900 m. In contrast, P. takahasii, occurs allopatrically around the Xingkai Lake in moist habitats. There is general agreement that both P. funebris and P. takahasii are intermediate to P. sylvestris var. mongolica and P. densiflora in some morphological traits (Table 1; Takenouchi, 1942; Mirov, 1967; Cheng and Fu, 1978; Fu et al., 1999). In the past, some investigators have placed them as different forms of either P. densiflora (for example, Takenouchi, 1942) or P. sylvestris (Cheng and Fu, 1978) rather than considering them as distinct species. However, both are phenetically and ecologically different from P. sylvestris var. mongolica and P. densiflora (Takenouchi, 1942; Cheng and Fu, 1978; Fu et al., 1999) and have been treated as distinct species by some taxonomists. Szmidt and Wang (1993), although not treating P. funebris as a distinct species, previously showed that plants of this type contained a mixure of allozyme polymorphisms from P. sylvestris var. mongolica and P. densiflora.
Pinus species exhibit paternal cp inheritance transmitted via pollen and maternal mt inheritance via seeds (Song et al., 2003; Chen et al., 2008; Zhou et al., 2010). This independent inheritance of two cytoplasmic genomes provides the opportunity for tracing paternal and maternal genetic changes within a single species because of different rates of gene flow and for discriminating female and male parents of hybrids (Ennos, 1994; Liston et al., 2007; Wachowiak and Prus-Głowacki, 2008). In addition, the analysis of multiple nuclear loci can be used to examine interspecific divergence and clarify parental origins of putative hybrid species (for example, Ma et al., 2006; Li et al., 2010). Thus, here we use sequences from cpDNA, mtDNA and eight nuclear genes to address the following specific questions. (1) Are P. sylvestris var. mongolica and P. densiflora clearly divergent for cp-, mt- and nuclear DNA? (2) How did these two widely distributed species in Northeast China respond to Quaternary climatic oscillations in terms of range shifts? (3) Is it possible that diploid P. funebris and P. takahasii originated following interspecific hybridization between P. sylvestris var. mongolica and P. densiflora?
Materials and methods
Population sampling
Needles were collected from 489 individuals across 47 natural populations throughout the natural distribution ranges of P. sylvestris var. mongolica, P. densiflora, P. funebris and P. takahasii (Figure 1, Supplementary Table S1). At four sites, P. sylvestris var. mongolica and P. densiflora occurred sympatrically (that is, at sites where populations 20–23 of P. sylvestris var. mongolica and populations 24–27 of P. densiflora co-occurred). Populations of the two possible hybrid species occurred allopatrically in the eastern or western Changbai Mountains and because of their restricted distributions only two and four populations of P. funebris and P. takahasii were sampled, respectively. In all, 7 to 12 individuals were sampled in each population, making sure that all individuals sampled were at least 100 m apart. We further used seeds from 78 trees of 18 populations (four to six individuals sampled per population, Supplementary Table S1) for sequencing nuclear loci. The latitude, longitude and altitude of each location sampled were measured by Extrex GIS (German, Taiwan). All needle samples were dried and stored in silica gel after collection. Seeds were stored at −20 °C, and soaked overnight in water at room temperature, before isolation of the haploid megagametophyte (the maternal tissue surrounding the embryo in gymnosperm seeds).
DNA extraction, amplification and sequencing
Total genomic DNA was isolated from 255 P. sylvestris var. mongolica, 148 P. densiflora, 37 P. funebris and 49 P. takahasii individuals, using approximately 20 mg of silica-gel dried, leaf-needle material per sample according to a hexadecetyltrimethyl ammonium bromide procedure (Doyle and Doyle, 1987) modified for use with an electric tissue homogenizer (QIAGEN, manufactured by Retsch, Qiagen, Valencia, CA, USA). Similarly, megagametophytes of seeds were separated and total DNA was extracted for sequencing nuclear loci. These megagametophytes were regarded as random gamete samples from each population. A total of 78 haploid DNA genomes from megagametophytes were analyzed for sequencing nuclear loci.
Three cpDNA regions: rpl16F71-rpl16R15, trnS-trnG and rbcL, were amplified and sequenced following the methods described in Zhou et al. (2010). For mtDNA, the nad4/3–4, nad5 intron 1 (Chen et al., 2008), nad1 intron B/C and nad7 intron 1 (Naydenov et al., 2007) regions that were found to be polymorphic in P. tabulaeformis and P. sylvestris, were surveyed for variation. Finally, for nuclear DNA we initially screened 20 loci across the four species and selected for further analysis only those loci represented by a single band generated by polymerase chain reaction (PCR). To check that such bands comprised a single sequence each PCR product was cloned into a pGEM T-easy vector (Promega Inc., Madison, WI, USA) and sequenced. Ten clones were sequenced per band. From the original 20 loci, eight (a3ip2, ccoaomt, pod, c3h-1, CesA2, dhn1, erd3 and aqua-MIP) were selected for inclusion in the total survey. PCR primers and the putative function of these eight loci are listed in Supplementary Table S2. All PCRs were conducted in a 25-μl volume, containing 10–40 ng plant DNA, 50 mm Tris-HCI, 1.5 mm MgCl2, 250 μg ml–1 bovine serum albumin, 0.5 mm dNTPs, 2 μM of each primer and 0.75 U of Taq polymerase. PCR products were purified using a TIANquick Midi Purification Kit following the recommended protocol (TIANGEN, Beijing, China).
To obtain the complete nucleotide sequence of the nad1 and rbcL regions, two to four different sequencing reactions were required using the PCR primers listed plus a pair of internal primers (nad1F1: 5′-TATTTCATAGGCAGCAGGTC-3′, nad1R1: 5′-GGGTCTTGGAGCGAACTACTC-3′) designed from the known sequence in P. sylvestris (Naydenov et al., 2007). These reactions were conducted on eight individuals per species, and then based on the complete sequences, two sequencing primers (nad1-R2: 5′-TCGCCCGACCTAAGAAGG-3′ and rbcL-F1: 5′-AGGCTGAGACGGGTGA-3′) were designed to sequence other trees included in the survey. All sequencing reactions used the ABI PRISM BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). DNA sequences were aligned with Clustal X and double checked by eye. All haplotype sequences are available from GenBank (accessions JF701446–JF701600).
Population genetic analysis
We used NETWORK version 4.2.0.1 to construct phylogenetic relationships among mtDNA, cpDNA and nuclear haplotypes (Bandelt et al., 1999; available at http://www.fluxus-engineering.com). Average mtDNA and cpDNA gene diversity within populations (HS), total gene diversity (HT), and the coefficients of differentiation GST and NST were estimated for each species except P. funebris, which comprised less than three populations, using PERMUT (available at http://www.pierroton.inra.fr/genetics/labo/Software/Permut/). We also compared the two estimates of population divergence, GST (coefficient of genetic variation over all populations; Nei, 1973) and NST (coefficient of genetic variation influenced by both haplotype frequencies and genetic distances between haplotypes), using a permutation test with 1000 permutations. We further examined Hierarchical partitioning of diversity among species, populations and individuals by analysis of molecular variance using ARLEQUIN version 3.0 (Excoffier et al., 2005), with significance tests based on 1000 permutations. As hybridization can result in recombination between some chlorotypes or mitotypes (Jaramillo-Correa and Bousquet, 2005), we used the four-gametic criterion defined by Hudson and Kaplan (1985) to infer possible recombination of cpDNA and mtDNA haplotypes. In doing this, we compared every pair of polymorphic nucleotide sites, for all four possible combinations of alleles (that is, Ab, AB, aB and ab) between the AB and ab haplotypes observed.
For nuclear sequences, we used DnaSP version 5.00.04 (Librado and Rozas, 2009) to estimate the number of segregating sites (S); Watterson's parameter (θW) (Watterson, 1975); nucleotide diversity (π) (Tajima, 1983), the minimum number of recombinant events (Rm) (Hudson and Kaplan, 1985) and net sequence divergence (Da) (Nei, 1987). We also calculated Tajima's D (Tajima, 1989), Fu and Li's D* and F* statistics (Fu and Li, 1993) and Fay and Wu's H (Fay and Wu, 2000) using the DnaSP version 5.00.04 software (Librado and Rozas, 2009). We used P._ponderosa, P._taeda, P._radiata and P._koraiensis as outgroups for Fay and Wu's H neutrality tests. In addition, Wright's fixation index, FST (Wright, 1951) was calculated for each locus and tested for significance by 1000 permutations as implemented in ARLEQUIN version 3.0 (Excoffier et al., 2005). Indels were excluded from all calculations other than for FST.
We also used STRUCTURE V. 2.3 (Hubisz et al., 2009) to assess the correspondence between species grouping and nuclear genotypic clustering. We assessed the likelihood of each number of clusters, K, (1⩽K⩽10) with allowance made for the correlation of allele frequencies between clusters. We performed 10 runs with a burn-in of 100 000 and then 500 000 iterations. We used Distruct v.1.1 to produce graphics (Rosenberg, 2004) and estimated the most likely number of clusters using the original method from Pritchard et al. (2000), and the ΔK statistics given in Evanno et al. (2005).
We used LAMARC v2.1.5 a coalescent-based method to estimate migrate rates (M) among the four species based on eight nuclear loci (Kuhner, 2006). This analysis used the Bayesian method with replication of chains and adaptive heating (Metropolis-Coupled Markov Chain Monte Carlo). Chains were repeated with different initial genealogies with each chain split into multiple searches, allowing for better sampling of parameter space. We used three independent MCMC runs of varying length and burn-in. All MCMC runs produced similar results, and therefore only results for the longest runs were presented, which were composed of three replicates of 10 initial chains and 2 long final chains. The initial chains were performed with 5000 samples and a sampling interval of 30 (150 000 steps), using a burn-in of 10 000 samples for each chain. The two final chains were carried out with the same burn-in and interval sampling, but with 50 000 samples (1 500 000 steps).
Tests for rapid expansion based on cpDNA sequences
Mismatch distributions of the observed number of nucleotide differences between pairs of cpDNA sequences were computed using the program Arlequin version 3.0. (Excoffier et al., 2005) to detect historical expansions in P. sylvestris var. mongolica and P. densiflora, for which ‘star-like’ phylogenies of rare chlorotypes were exhibited. We assumed that populations at demographic equilibrium would have a multimodal distribution of pairwise differences, while those that experienced a sudden demographic expansion should display a unimodal distribution (Slatkin and Hudson, 1991). We tested three data sets: (I) all samples of P. sylvestris var. mongolica, (II) all samples of P. densiflora and (III) samples with chlorotypes specific to P. densiflora. We used a total of 1000 parametric bootstrap replicates based on segregating sites to generate an expected distribution under a model of sudden demographic expansion (Rogers and Harpending, 1992). We also used the sum of squared deviations (SSD) as a statistic to test the validity of the expansion model with P-values calculated as the proportion of simulations producing a larger SSD than the observed SSD. We calculated the raggedness index and its significance to quantify the smoothness of the observed mismatch distribution.
For each of the same three groups (I–III), we also calculated Fu's FS value, which is primarily based on differences between expected numbers of alleles (estimated through 10 000 computer simulations based on the observed pairwise differences in our sample) and observed numbers of alleles. This statistic is very sensitive to a recent demographic expansion for which large, negative values are typically obtained (Fu, 1997). Values of Tajima's D (Tajima, 1989) were also calculated to assess population expansion (using the total number of mutations), for which negative values are also expected. Estimation and testing were conducted using Arlequin version 3.0 (Excoffier et al., 2005) with 1000 bootstrap replicates for both Fu's FS and Tajima's D.
To further assess the demographic history of species, we also used LAMARC v2.1.5 (Kuhner, 2006), which takes account of genealogical relationships among haplotypes, to estimate the exponential population growth rate parameter g. For the three groups (I–III), we also conducted three independent MCMC runs as detailed above. All MCMC runs produced similar results. Large and positive values of the exponential growth parameter g indicate population expansion, whereas negative values indicate population shrinkage. The scale of g is known to be biased upward, and thus relatively small positive values (g=10) may indicate little or no growth, whereas small negative values (g=−10) may indicate important population size declines (LAMARC documentation: http://evolution.gs.washington.edu/lamarc/).
Tests of alternative scenarios for the origins of P. funebris and P. takahasii by ABC modeling
To obtain a more detailed inference on the evolutionary histories of these four species, we tested potential scenarios using the approximate Bayesian computation procedure (ABC) in DIYABC v1.0.4.39 (Cornuet et al., 2008, 2010) based on eight nuclear loci. Data can be simulated under any number of scenarios of population divergence, population size change and admixture (Cornuet et al., 2008). We tested three possible origin scenarios of P. funebris and P. takahasii, respectively: admixture from both putative parental species, divergence only from P. sylvestris var. mongolica and divergence only from P. densiflora. Based on the cpDNA analysis, both P. sylvestris var. mongolica and P. densiflora experienced rapid range expansions. So, we added population size change models to these three scenarios (Supplementary Figure S1). For the ABC analysis, parameter values were set from the minimum to maximum range of priors. We chose the number of haplotypes, number of segregating sites and mean pairwise difference as one-sample summary statistics, and the mean of pairwise differences (W) and (B) as two-sample summary statistics to compare observed and simulated data sets. A reference table consisting of 1 000 000 simulated data sets per scenario was created. We used 1% of the simulated data sets closest to the observed data to estimate the relative posterior probability (with 95% confidence intervals (CIs)) of each scenario via a logistic regression and posterior parameter distributions according to the most likely scenario (Cornuet et al., 2008, 2010). We assumed a generation time of 25 years for each of the four species as suggested for other pine species (Brown et al., 2004) and our field observations.
Results
cpDNA variation
Based on sequence variation exhibited in the three cpDNA fragments (rpl16, trnS-trnG, rbcL) analysed, 13 chlorotypes (Supplementary Table S3) were identified across all individuals of the four species sampled with 2 chlorotypes recorded as frequent (C1 and C8) and 11 as relatively rare (C2–C7, C9–C13) (Figure 1a; Supplementary Table S1). Whereas P. sylvestris var. mongolica possessed chlorotype C1 at high frequency (94.5%), P. densiflora was characterized by a high frequency of chlorotype C8 (88.5%). Chlorotype C1 was fixed in 12 of 23 populations surveyed in P. sylvestris var. mongolica and occurred at high frequency in the remaining populations. In the same species, chlorotypes C2–C4 and C7 were present at low frequency in northwestern populations, while C5 and C6 occurred only in eastern populations that were sympatric with P. densiflora. Three of the chlorotypes found in P. sylvestris var. mongolica were also recorded at very low frequency in P. densiflora, that is, C1 in populations 25–28, C2 in population 24, and C5 in population 29. In P. densiflora, 7 of the 18 populations surveyed were fixed for chlorotype C8, while the remaining populations contained C8 at high frequency. Some populations of P. densiflora also contained at low frequency chlorotypes C9–C11, which along with C8 were never recorded in P. sylvestris. The two putative hybrid species, P. funebris and P. takahasii, contained a mixture of chlorotypes from both parent species. Thus, chlorotypes C1, C2, C4 and C8 were present in P. funebris, while C1, C3, C8 and C9 occurred in P. takahasii. In addition, each putative hybrid species contained one species-specific chlorotype at low frequency—C12 being specific to P. funebris and C13 being specific to P. takahasii.
The minimum-spanning network for chlorotypes (Figure 1b) identified two distinct groups, which mostly represent chlorotypes possessed by the two parental species, respectively. The putative hybrid species possessed chlorotypes from both groups. Values for average within-population cpDNA diversity (0.535) and total genetic diversity (0.607) in P. takahasii were much higher than in P. sylvestris var. mongolica (0.109, 0.109) and P. densiflora (0.196, 0.201). NST and GST were low in both P. densiflora and P. sylvestris var. mongolica, and in neither case was NST significantly larger than GST (Supplementary Table S4). Analysis of molecular variance showed that (77.08%) of the total cpDNA variation was distributed between species, and that variation within species, was mostly due to that present within populations. Analysis of molecular variance conducted only on the putative parental species showed that 91.20% of the variation was distributed between them (Supplementary Table S5).
mtDNA variation
A combined analysis of mutations and indels across the four mtDNA fragments surveyed (nad4/3–4, nad5 intron 1, nad1 intron B/C and nad7 intron 1) identified six different mitotypes (Supplementary Table S3), M1–M6 (Figure 1c; Supplementary Table S1) over all trees examined. In P. sylvestris var. mongolica, all populations except two were fixed for M1 and the overall frequency of M1 in the species was 99.2%. The two other mitotypes found in this species occurred in single individuals within different populations, M5 in population 22 and M6 in population 23. In P. densiflora, a strong geographical pattern for mtDNA variation was evident. Northern populations of this species (populations 24–27) were fixed for mitotype M3 (overall frequency in species equalled 34.5%), while southern populations (32–41) were fixed for mitotype M2 (overall frequency in species equaled 58.8%). In contrast, central populations sampled from Jilin province (populations 28–31) were fixed for either M2 (population 30) or M3 (population 28), or were polymorphic for M2 and M4 (populations 29 and 31). Mitotypes M3 and M4, which differed from M2 by a single-nucleotide substitution, occurred at very high frequency in P. takahasii and P. funebris, respectively. None of the mitotypes found in P. sylvestris var. mongolica (M1, M5 and M6) was recorded in either putative hybrid species.
The mitotype network (Figure 1d) indicated that mitotypes found in P. sylvestris var. mongolica formed one group while those present in the two putative hybrid species and P. densiflora formed another group. Total mtDNA diversity was much higher in P. densiflora (HT=0.522) than in P. sylvestris var. mongolica (HT=0.028) and P. takahasii (HT=0.020), while average within-population diversity was similar (HS=0.028 in P. sylvestris var. mongolica, HS=0.046 in P. densiflora, and HS=0.029 in P. takahasii). In P. densiflora, NST (0.919) was not significantly greater than GST (0.913) (P>0.05, Supplementary Table S4). As expected, analysis of molecular variance showed that most variation was distributed between species (97.55%) while variation between populations within species was significant only in P. densiflora (Supplementary Table S5).
Nuclear loci
The amount of nucleotide polymorphism in each species varied greatly among loci with the dhn1 locus being most polymorphic in all four species (Supplementary Table S6). Average estimates of polymorphism over the eight loci were similar across species with silent polymorphism being three- to fivefold greater than polymorphism at nonsynonymous sites. P. funebris and P. takahasii exhibited similar levels of silent nucleotide diversity that were somewhat higher than those exhibited by P. sylvestris var. mongolica and P. densiflora (Table 2).
FST and net sequence divergence (Da) were calculated to measure genetic differentiation among species at each locus (Table 3). Results for Da were similar to FST and thus are not presented here. FST-values were mostly significantly different from zero at each locus between P. sylvestris var. mongolica and one or other of the other three species. Significant FST-values were also detected at the a3ip2, ccoaomt and dhn1 loci between P. densiflora and P. funebris, at the ccoaomt, dhn1 and erd3 loci between P. densiflora and P. takahasii and at the pod and CesA2 loci between P. funebris and P. takahasii, respectively (Table 3). The genetic differentiation was much smaller between P. densiflora and P. funebris and P. takahasii, as compared with P. sylvestris var. mongolica and the latter two species.
We calculated Tajima's D, Fu and Li's D* and F* and Fay and Wu’s H to detect departures from the standard neutral model of molecular evolution at each locus. Fu and Li's D* tests showed a similar trend to F* (data not shown). Most of the tests did not significantly deviate from the standard neutral model (Table 4). We detected significant negative D or H at the ccoaomt locus in P. sylvestris var. mongolica and P. densiflora, and significant negative D and D* at the CesA2 locus in P. sylvestris var. mongolica. No significant value was detected in either putative hybrid species.
Haplotype genealogies constructed for each locus by NETWORK (Figure 2) could be grouped into three different types. One type exhibited by the a3ip2 and ccoaomt loci showed a high level of haplotype distinction between P. sylvestris var. mongolica and P. densiflora. Thus, 7 of the 12 haplotypes detected at the a3ip2 locus were specific to P. sylvestris var. mongolica, while 3 were specific to P. densiflora. Similarly, at the ccoaomt locus, two of the 15 haplotypes detected were specific to P. sylvestris var. mongolica while 9 were specific to P. densiflora. At neither locus were any haplotypes shared between these two taxa. All haplotypes possessed by P. sylvestris var. mongolica were separated from those possessed by P. densiflora by at least five evolutionary steps (mutations) based on most parsimonious relationships. The putative hybrid species, P. takahasii and P. funebris, possessed a mixture of haplotypes specific to P. sylvestris var. mongolica and P. densiflora as well as some haplotypes (alleles) specific to themselves.

Haplotype genealogies for eight nuclear loci. The color of each sector of a circle indicates the frequency of a haplotype recorded in each pine species. The size of a circle is proportional to the frequency of a haplotype across the four pines. Branch lengths longer than one mutation step are marked on each branch.
The second type of genealogy was observed for five loci (pod, c3h-1, CesA2, dhn1 and erd3) where one central haplotype shared by P. sylvestris var. mongolica, P. densiflora and at least one of the two putative hybrid species was ancestral to other haplotypes. In this genealogy, both P. takahasii and P. funebris also shared a few derived haplotypes that were specific to either P. sylvestris var. mongolica or P. densiflora, as well as possessing a few haplotypes not found in either putative parent species. In the final class of haplotype network exhibited for the aqua-MIP locus, the most common haplotypes were mainly found in either P. sylvestris var. mongolica or P. densiflora and one or both putative hybrid species. The one exception to this was for H2, which was present in all four taxa. Once again, a few haplotypes were recorded in P. takahasii and P. funebris that were not present in either putative parent species. Despite the different types of genealogies noted across the eight loci, two common features were evident: (1) haplotypes specific to either P. sylvestris var. mongolica or P. densiflora were commonly shared by P. takahasii and P. funebris, and (2) some haplotypes were specific to the putative hybrid species (that is, 10 haplotypes were specific to P. funebris and 14 to P. takahasii).
We also used the Bayesian clustering algorithm implemented in the program STRUCTURE to detect genetic admixture of the two possible hybrid species. Model selection based on the ΔK criterion (Evanno et al., 2005) supported K=2. Values of LnPD showed an increase with increasing K using the original method of Pritchard et al. (2000), although the increase was very low when K⩾6. The STRUCTURE clustering results for K=2–6 are presented in Supplementary Figure S2). When K=2, both P. funebris and P. takahasii were shown to comprise a genetic mixture of P. densiflora and P. sylvestris var. mongolica, with more of their ancestry coming from the former species.
Coalescent-based estimates from LAMARC indicated that the input of polymorphism to the assumed hybrid species from their putative parental species following historical migration was very high relative to the mutation rate (M=m/μ). The most probable estimates of M ranged from 158 to 619, with the highest migration estimated from P. densiflora into P. funebris and P. takahasii (M=619–573) (Supplementary Table S7).
Demographic expansion analysis based on cpDNA sequences
The mismatch distribution for all chlorotypes recorded in P. sylvestris var. mongolica (group I) showed a unimodal pattern (Figure 3a), which is indicative of a past range expansion in this species. In P. densiflora (group II), two peaks were observed in the mismatch distribution (Figure 3b), which might reflect introgression of some chlorotypes from P. sylvestris var. mongolica to P. densiflora. This possibility is supported by the fact that a unimodal mismatch distribution was obtained when chlorotypes shared with P. sylvestris were excluded from analysis (group III, Figure 3c). Thus, a past range expansion might also be inferred for P. densiflora. Further analyses of the variance (SSD) and raggedness index suggested that the curves did not differ significantly from those of distributions expected from a model of sudden population expansion (Table 5).

Results of the mismatch distribution analysis for (a) all chlorotypes recorded in P. sylvestris var. mongolica, (b) all chlorotypes recorded in P. densiflora and (c) chlorotypes specific to P. densiflora. Continuous lines show the distributions expected for an expanding population, while the dotted lines represent the observed distributions of pairwise differences among samples.
Sudden and recent range expansions of both P. sylvestris var. mongolica and P. densiflora in Northeast China are also supported by significant negative values obtained for FS and Tajima's D values in chlorotype groups I and III, and by large values obtained for the exponential population growth rate parameter ‘g’ derived by LAMARC tests (Kuhner, 2006) on all three groups of cpDNA sequences, groups I–III (Table 5).
Favored scenarios for the origins of P. funebris and P. takahasii based on ABC simulations
Posterior probabilities clearly favored the admixture origin hypothesis for both P. funebris and P. takahasii (Table 6, Supplementary Figure S1, 3, Supplementary Tables S8, S9, S10). More specifically, our ABC simulations that included models of changes in effective population size supported the hypothesis that P. sylvestris var. mongolica and P. densiflora expanded in the past. P. sylvestris var. mongolica was estimated to expand around 190 (95% CI: 68–248) thousand years ago (kya) and P. densiflora about 110 (95% CI: 23–220) kya. In addition, the posterior parameter estimations suggested that the admixture origins of P. funebris and P. takahasii from P. sylvestris var. mongolica and P. densiflora occurred around 33 (median, 95% CI: 2–73) kya and 35 (median, 95% CI: 2–73) kya, respectively. The two parental species were estimated to have diverged from their most recent common ancestor (MRCA) around 1.85 million years ago.
Discussion
Our comparison of maternally inherited mtDNA, paternally inherited cpDNA and biparentaly inherited nuclear DNA sequence variation within and between four closely related pine species in Northeast China, revealed that the two widespread species, P. sylvestris var. mongolica and P. densiflora, were genetically diverged and delimited by mtDNA, cpDNA and two of eight nuclear genes examined. In areas of sympatry, some hybrids between these two species were identified based on chlorotype and nuclear genotype composition. Population genetic analysis suggested that both species had experienced rapid range expansion during the late Quaternary. This we propose led to secondary contact between them and subsequent interspecific hybridization, which is possibly resulted in the homoploid hybrid origin of the narrowly distributed pines, P. funebris and P. takahasii, each of which possesses a mixture of cpDNA and nuclear haplotypes that distinguish P. sylvestris var. mongolica from P. densiflora.
Genetic divergences between P. densiflora and P. sylvestris var. mongolica
Closely related species are often divergent at some loci, but share alleles at other loci (Nosil et al., 2009). Sharing of alleles may result from incomplete lineage sorting and/or interspecific introgression (Gow et al., 2006). P. densiflora and P. sylvestris var. mongolica differed in mitotype composition (Figure 1; Supplementary Table S1) with P. densiflora containing three closely related mitotypes (M2, M3 and M4), while P. sylvestris contained the distantly related M1 mitotype at very high frequency plus two very rare mitotypes, M5 an M6. According to the ‘four gametic criterion’, mitotypes M5 and M6 were assumed to be recombinants of M1 and M2 (Hudson and Kaplan, 1985; Jaramillo-Correa and Bousquet, 2005), which is consistent with their distribution only in areas of sympatry with P. densiflora.
P. densiflora and P. sylvestris var. mongolica were also largely distinguished by the chlorotypes they possessed with chlorotypes C8 and C1 occurring at high frequency in P. densiflora (88.5%) and P. sylvestris (94.5%), respectively. These two chlorotypes were separated by two steps in a chlorotype network in which they were located centrally (Figure 1). The remaining 11 chorotypes identified in the analysis were rare and according to the network were derived from one or other of the two common chlorotypes. Three chlorotypes (C1, C2 and C5) found in P. sylvestris var. mongolica were present at low frequency in sympatric populations of P. densiflora, indicating recent cryptic introgression.
Taken together, our analysis of mtDNA and cpDNA variation showed that the two widespread species investigated, P. densiflora and P. sylvestris var. mongolica, can be largely delimited by both the mtDNA and cpDNA haplotypes they possess, although a low level of chlorotype sharing occurs in areas of sympatry. The finding for mtDNA contrasts with those of some previous surveys of variation in coniferous species, which showed a high degree of mitotype sharing across closely related species and an inability to delimit species according to mitotype (Du et al., 2009; Zhou et al., 2010).
The two widespread pine species were also distinguished by different nuclear haplotypes at two of eight nuclear loci surveyed (a3ip2 and ccoaomt), and FST values (Table 3) were significant at these loci; however, at the remaining six loci there was considerable haplotype sharing between the species (Figure 2). Significant negative Tajima's D or Fay and Wu’s H were only observed at the ccoaomt locus in P. sylvestris var. mongolica and P. densiflora, indicating a higher signature of selection or demographic expansion at this locus. The a3ip2 gene encodes a protein that interacts with the protein ABI3 that possibly functions in the control of bud growth and flowering (Palmé et al., 2009), while the ccoaomt gene encodes a key enzyme for lignin biosynthesis involved in plant defense (Almagro et al., 2009). Allelic substitutions at both genes may have had important roles in the adaptive divergence and reproductive isolations of the two species. Thus, complete lineage sorting at these two loci might be due to their linkage to speciation genes (hitchhiking) or to their direct role in adaptive divergence. If this is correct, the two genes would be expected to have been subjected to strong selection-driven lineage sorting during speciation and also following any subsequent introgression events (Via, 2009). The shared haplotypes (alleles) at the remaining six nuclear loci examined, which were located at central positions in the haplotype networks (Figure 2), may derive from ancestral polymorphisms, whereas those positioned at the tips of branches are more likely due to introgression between the two species (Zheng and Ge, 2010). If these genes are not linked to speciation genes or have little direct role in the adaptive divergence of the two species, lineage sorting of ancestral polymorphisms and re-sorting of introgressed alleles at these loci would be expected to occur more slowly (Ting et al., 2000) and therefore explain the haplotype sharing observed.
Glacial refugia and range expansions of P. densiflora and P. sylvestris
The three mitotypes recorded within P. densiflora differed in their geographical distribution although NST was not significantly greater than GST (Supplementary Table S6). Whereas mitotype M2 was widely distributed in the southern part of Northeast China, M3 was fixed in multiple populations sampled from the Changbai Mountains to the north. This geographical pattern of variation suggests that the species’ range may have been fragmented in the past into at least two isolated glacial refugia located in the northeast and south (Supplementary Figure S4a), respectively. This is of interest as studies of other forest trees have also indicated the occurrence of more than one glacial refugium for forests in northeast Asia (for example, Aizawa et al., 2007; Bai et al., 2010).
In contrast to the situation for mitotype variation, all populations of P. densiflora shared a common chlorotype (C8). Such uniformity could have been caused by the homogenizing effect of a high level of gene flow through long-distance dispersed pollen preventing cpDNA divergence occurring among populations in geographically isolated glacial refugia (Supplementary Figure S4a). The occurrence of a ‘star-like’ phylogeny of rare chlorotypes derived from chlorotype C8 (Figure 1) indicates that an extensive expansion in the geographical range of P. densiflora occurred in the recent past. Similarly, the presence of one widespread mitotype in P. sylvestris var. mongolica together with a ‘star-like’ phylogeny of rare chlorotypes derived from the most common chlorotype (C1) suggests an extensive range expansion of this species in Northeast China in the relatively recent past (Supplementary Figure S4b). The latter is consistent with the findings of a recent phylogeographic study of P. sylvestris over its Eurasian distribution, which showed that central Asia was one of its glacial refugia from where the species colonized Northeast China recently and expanded there (Naydenov et al., 2007).
The occurrence of recent range expansions of P. densiflora and P. sylvestris var. mongolica were also supported by the significant negative values obtained for Fu's FS and Tajima's D (Table 5), the demonstration of unimodal mismatch distributions, and the large population expansion rates (g>500; Table 5) based on cpDNA sequence data, plus the effective population size change models estimated by ABC based on eight nuclear loci. Neutrality tests of nuclear data partly supported a recent expansion hypothesis for both species in that the estimated values of Tajima's D and Fu and Li's D* were mostly negative; however, because of limited sample size, departures from the standard neutral model of molecular evolution were not statistically significant (Table 4). The range expansions of P. sylvestris var. mongolica and P. densiflora were estimated to have occurred about 190 and 110 kya, respectively, based on nuclear data (Supplementary Tables S9–10). These dates suggest that the Last Glacial Maximum (∼20 kya) probably had little effect on the range contractions and expansions of these two species in the late Quaternary. Instead, the dates are more consistent with the period following the largest Quaternary glaciation recorded for the Asian mountains, which began approximately 800 kya and continued until 200 kya (Zheng et al., 2002; Wu et al., 2010).
It is feasible that during this glaciation, the range of P. densiflora was fragmented into different refugia in Northeast China, and following the end of this glacial period it expanded its range both southward and northeastward. At the same time, P. sylvestris is believed to have colonized Northeast China from central Asia (Naydenov et al., 2007), and after expanding its range southward will have come into contact and hybridized with P. densiflora (Supplementary Figures S4b and c).
Hybrid origins of P. funebris and P. takahasii?
Evidence supporting the hypothesis that both P. funebris and P. takahasii are possibly homoploid hybrid species emerged from the analysis of cp, mt and nuclear DNA variation. Both species contained cp and nuclear haplotypes that distinguished the two putative parent species, P. densiflora and P. sylvestris var. mongolica, and STRUCTURE confirmed that they were a genetic mixture of the two widespread species but with a greater proportion of their nuclear ancestry coming from P. densiflora (Table 3, Supplementary Figure S3). This is consistent with the results of a previous study based on allozyme polymorphisms (Szmidt and Wang, 1993). Moreover, the most favored scenarios that emerged from the ABC simulations conducted strongly supported an admixed origin of P. funebris and P. takahasii from the other two species approximately 33–35 kya. Both putative hybrid species were almost fixed for different mitotypes only found in P. densiflora, indicating that different individuals of P. densiflora had served as the maternal parents of P. funebris and P. takahasii, respectively.
These two putative hybrid species are likely to be reproductively isolated from their parents through spatial/habitat isolation. P. funebris grows at higher elevations relative to either parent in the Changbai Mountains, while P. takahasii occurs in wetter habitats around the Xingkai Lake (Cheng and Fu, 1978) from which both putative parents are excluded. Such ecological differentiation from parental taxa appears very common for most recorded homoploid hybrid species (Gross and Rieseberg, 2005). For example, in pines, P. densata, which is derived from hybridization between P. yunnanensis and P. tabulaeformis, inhabits a high mountain environment where the parental species are apparently unable to grow (Wang et al., 2001). It has also been reported that whereas P. densiflora sheds its pollen in April, both putative hybrid species and P. sylvestris var. mongolica release their pollen from May to June (Cheng and Fu, 1978). This difference in pollen shedding time might therefore also contribute to reproductive isolation between the two putative hybrid species and P. densiflora, although this possibility will need to be verified by detailed analysis.
Although one scenario for the origin of P. funebris and P. takahasii is homoploid hybrid speciation, an alternative scenario that cannot be discounted is that both taxa originated by divergence (for example, from P. densiflora), possibly as a result of adaptation to different habitats, and subsequently were introgressed (for example, by P. sylvestris var. mongolica) (see Wachowiak et al., 2011, for a possible example of divergence followed by introgression in other pine species). In this case, their hybrid nature might be coincidental and not directly responsible for their origin, in that the introgressed genes may have had no part in their reproductive isolation from their parents. Deciding between these two evolutionary scenarios, that is, homoploid hybrid speciation or divergence from a parent species followed by introgression, is difficult. If in future it is shown that genes contributed by both parent species were instrumental in causing the reproductive isolation of P. funebris and P. takahasii, for example by enabling a shift in habitat, then divergence following hybridization might be assumed and thus both species could be concluded to be homoploid hybrid species. Until then, however, we should continue to consider P. funebris and P. takahasii as putative hybrid species rather than as fully proven hybrid species.
The finding that only P. densiflora served as the maternal parent of both putative hybrid species implies mainly unidirectional hybridization between P. sylvestris var. mongolica and P. densiflora in the wild as reported to have occurred between some other pine species (Watano et al., 2004; Liston et al., 2007; Wachowiak and Prus-Głowacki, 2008). Such unidirectional hybridization might have two alternative causes. First, it is feasible that P. densiflora stops shedding pollen before the ovules of P. sylvestris var. mongolica become receptive, whereas the later developed ovules of P. densiflora may be available to early-shed pollen of P. sylvestris. Second, hybrid survival may be species-biased (Currat et al., 2008) with only those having P. densiflora as a maternal parent being viable. This has been reported to be the case for some other hybrids between other pine species (Wachowiak et al., 2005), that is, hybrids mothered by one species are viable, but are inviable if produced by the reciprocal cross.
Conclusion
Our genetic analysis has shown that P. funebris and P. takahasii contain highly diverged chlorotypes and nuclear alleles that distinguish P. sylvestris var. mongolica and P. densiflora. During the most severe glacial stage of the Quaternary, P. sylvestris and P. densiflora probably remained allopatric: the former species surviving in glacial refugia in central Asia and Europe (Naydenov et al., 2007) while the latter was restricted to two or more refugia in Northeast China. At the end of this period, P. sylvestris var. mongolica is thought to have colonized Northeast China and expanded its range southward into the distribution range of P. densiflora, which had expanded its range northeastward. Such contact lead to interspecific hybridization and the possible origin of two putative homoploid hybrid species, P. funebris and P. takahasii. Although our findings based on detailed analysis of large population genetic data sets are consistent with the homoploid hybrid origin of these two species, we are unable to exclude fully other possible origins for them. For example, an origin by divergence from P. densiflora followed by introgression from P. sylvestris var. mongolica. This emphasizes the difficulty of confirming homoploid hybrid speciation from such data sets and the need to study where possible the process ‘in action’ to fully understand the mechanisms that drive it (Abbott et al., 2010).
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad.cb052n9j.
References
Abbott RJ (1992). Plant invasions, interspecific hybridization and the evolution of new plant taxa. Trends Ecol Evol 7: 401–405.
Abbott RJ, Brennan AC, Hegarty MJ, Hiscock SJ (2010). Homoploid hybrid speciation in action. Taxon 59: 1375–1386.
Abbott RJ, Brochmann C (2003). History and evolution of the arctic flora: in the footsteps of Eric Hultén. Mol Ecol 12: 299–313.
Aizawa M, Yoshimaru H, Saito H (2007). Phylogeography of a northeastern Asian spruce, Picea jezoensis, inferred from genetic variation observed in organelle DNA markers. Mol Ecol 16: 3393–3405.
Almagro L, Gómez Ros LV, Belchi-Navarro S, Bru R, Ros Barceló A, Pedreño MA (2009). Class III peroxidases inplant defence reactions. J Exp Bot 60: 377–390.
Arnold ML (2006). Evolution through Genetic Exchange. Oxford University Press: New York.
Bai WN, Liao WJ, Zhang DY (2010). Nuclear and chloroplast DNA phylogeography revealtwo refuge areas with asymmetrical gene flow in atemperate walnut tree from East Asia. New Phytol 188: 892–901.
Bandelt HJ, Forster P, Röhl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Brown G, Gill G, Kuntz R, Langley C, Neale D (2004). Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc Natl Acad Sci USA 101: 15255–15260.
Chen KM, Abbott RJ, Milne RI, Tian XM, Liu JQ (2008). Phylogeography of Pinus tabulaeformis Carr. (Pinaceae), a dominant species of coniferous forest in northern China. Mol Ecol 17: 4276–4288.
Cheng WC, Fu LG (1978). Chinese flora. Sci Press 7: 239–280. (in Chinese).
Cornuet JM, Ravigné A, Estoup A (2010). Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v. 1.0). BMC Bioinform 11: 401.
Cornuet JM, Santos F, Beaumont MA, Robert CP, Marin JM, Balding DJ et al. (2008). Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24: 2713–2719.
Currat M, Ruedi M, Petit RJ, Excoffier L (2008). The hidden side of invasions: massive introgression by local genes. Evolution 62: 1908–1920.
Doyle JJ, Doyle JL (1987). A rapid DNA isolation procedure for small quantities of fresh leaf material. Phytochem Bull 19: 11–15.
Du KF, Petit RJ, Liu JQ (2009). More introgression with less gene flow: chloroplast vs. mitochondrial DNA in the Picea asperata complex in china, and comparison with other conifers. Mol Ecol 18: 1396–1407.
Ennos R (1994). Estimating the relative rates of pollen and seed migration among plant populations. Heredity 72: 250–259.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S (2005). ARLEQUIN (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Fay JC, Wu CI (2000). Hitchhiking under) positive Darwinian selection. Genetics 131: 479–491.
Fu LG, Li N, Elias TS (1999). Pinaceae. In: Wu Z, Raven PH (eds). Flora of China Vol. 4. Science Press: Beijing. pp 11–52.
Fu YX (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 147: 915–925.
Fu YX, Li WH (1993). Statistical tests of neutrality of mutations. Genetics 133: 693–709.
Gow JL, Peichel CL, Taylor EB (2006). Contrasting hybridization rates between sympatric three-spined sticklebacks highlight the fragility of reproductive barriers between evolutionarily young species. Mol Ecol 15: 739–752.
Gross BL, Rieseberg LH (2005). Ecological genetics of homoploid hybrid speciation. J Hered 96: 241–252.
Hewitt GM (2000). The genetic legacy of the Quaternary ice ages. Nature 405: 907–913.
Hewitt GM (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B 359: 183–195.
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9: 1322–1332.
Hudson RR, Kaplan NL (1985). Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164.
Jaramillo-Correa JP, Bousquet J (2005). Mitochondrial genome recombination in the zone of contact between two hybridizing conifers. Genetics 171: 1951–1962.
Kim M, Cui ML, Cubas P, Gillies A, Lee K, Chapman MA et al. (2008). Regulatory genes control a key morphological and ecological trait transferred between species. Science 322: 1116–1119.
Kuhner MK (2006). LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22: 768–770.
Li Y, Stocks M, Hemmilä S, Källman T, Zhu HT, Zhou YF et al. (2010). Demographic histories of four spruce (Picea) species of the Qinghai-Tibetan Plateau and neighboring areas inferred from multiple nuclear loci. Mol Biol Evol 27: 1001–1014.
Librado P, Rozas J (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Liston AA, Parker-Defeniks M, Syring JV, Willyard A, Cronn R (2007). Interspecific phylogenetic analysis enhances intraspecific phylogeographical inference: a case study in Pinus lambertiana. Mol Ecol 16: 3926–3937.
Ma XF, Szmidt AE, Wang XR (2006). Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Mol Biol Evol 23: 807–816.
Mallet J (2007). Hybrid speciation. Nature 446: 279–283.
Mirov NT (1967). The Genus Pinus. Ronald Press: New York.
Naydenov K, Senneville S, Beaulieu J, Tremblay F, Bousquet J (2007). Glacial vicariance in Eurasia, mitochondrial DNA evidence from Scots pine for a complex heritage involving genetically distinct refugia at mid-northern latitudes and in Asia Minor. BMC Evol Biol 7: 233.
Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Nosil P, Funk DJ, Ortiz-Barrientos D (2009). Divergent selection and heterogeneous genomic divergence. Mol Ecol 18: 375–402.
Palmé AE, Pyhäjärvi T, Wachowiak W, Savolainen O (2009). Selection on nuclear genes in a Pinus phylogeny. Mol Biol Evol 26: 893–905.
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al. (2003). Glacial refugia: hotspot but not melting pots of genetic diversity. Science 300: 1563–1565.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rieseberg LH (1997). Hybrid origins of plant species. Annu Rev Ecol Syst 27: 259–389.
Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T et al. (2003). Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301: 1211–1216.
Rogers AR, Harpending H (1992). Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9: 552–569.
Rosenberg N (2004). DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Slatkin M, Hudson RR (1991). Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129: 555–562.
Song BH, Wang XQ, Wang XR, Ding KY, Hong DY (2003). Cytoplasmic composition in Pinusdensata and population establishment of the diploid hybrid pine. Mol Ecol 12: 2995–3001.
Song BH, Wang XQ, Wang XR, Sun LJ, Hong DY, Peng PH (2002). Maternal lineages of Pinus densata, a dipoid hybrid. Mol Ecol 11: 1057–1063.
Szmidt AE, Wang XR (1993). Molecular systematics and genetic differentiation of Pinus sylvestris (L.) and P. densiflora (Sieb. et Zucc.). Theor Appl Genet 86: 159–165.
Tajima F (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460.
Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Takenouchi M (1942). A preliminary report on the conifers of Manchukuo. J Jpn For Soc 24: 113–129.
Ting CT, Tsaru SC, Wu CI (2000). The phylogeny of closely related species as revealed by the genealogy of a speciation gene, Odysseus. Proc Natl Acad Sci USA 97: 5313–5316.
Via S (2009). Natural selection in action during speciation. Proc Natl Acad Sci USA 106: 9939–9946.
Wachowiak W, Lewandowski A, Prus-Głowacki W (2005). Reciprocal controlled crosses between Pinus sylvestris and P. mugo verified by a species-specific cpDNA marker. J Appl Genet 46: 41–43.
Wachowiak W, Palmé AE, Savolainen O (2011). Speciation history of three closely related pines Pinus mugo (T.), P. uliginosa (N.) and P. sylvestris (L.). Mol Ecol 20: 1729–1743.
Wachowiak W, Prus-Głowacki W (2008). Hybridisation processes in sympatric populations of pines Pinus sylvestris L., P. mugo Turra and P. uliginosa Neumann. Plant Syst Evol 271: 29–40.
Wang BS, Mao JF, Gao J, Zhao W, Wang XR (2011). Colonization of the Tibetan Plateau by the homoploid hybrid pine Pinus densata. Mol Ecol 20: 3796–3811.
Wang XR, Szmidt AE, Lewandowski A, Wang ZR (1990). Evolutionary analysis of Pinus densata Masters, a putative tertiary hybrid 1. Allozyme variation. Theor Appl Genet 80: 635–640.
Wang XR, Szmidt AE, Savolainen O (2001). Genetic composition and diploid hybrid speciation of a high mountain pine, Pinus densata, native to the Tibetan Plateau. Genetics 159: 337–346.
Wang XR, Tsumura Y, Yoshimaru H, Nagasaka K, Szmidt AE (1999). Phylogenetic relationships of Eurasian pines (Pinus, Pinaceae) based on chloroplast rbcL, matK, rpl20-rps18 spacer, and trnV intron sequences. Am J Bot 86: 1742–1753.
Watano Y, Kanai A, Tani N (2004). Genetic structure of hybrid zones between Pinus pumila and P. parviflora var. pentaphylla (Pinaceae) revealed by molecular hybrid index analysis. Am J Bot 91: 65–72.
Watterson GA (1975). On the number of segregating sites in the genetical models without recombination. Theor Popul Biol 7: 256–276.
Wright S (1951). The genetical structure of populations. Ann Eugen 15: 323–354.
Wu CY (1995). Vegetation of China 2nd edn Science Press: Beijing.
Wu LL, Cui XK, Milne RI, Sun YS, Liu JQ (2010). Multiple autopolyploidizations and range expansion of Allium przewalskianum Regel. (Alliaceae) in the Qinghai-Tibetan Plateau. Mol Ecol 19: 1691–1704.
Zhang HF, Li MX (1984). Study of karyotypes of three species of Pinus. J North-Eastern Forestry Inst 12: 115–121. (in Chinese).
Zheng B, Xu Q, Shen Y (2002). The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan plateau: review and speculation. Quatern Int 97–98: 93–101.
Zheng XM, Ge S (2010). Ecological divergence in the presence of gene flow in two closely related Oryza species (Oryza rufipogon and O. nivara). Mol Ecol 19: 2439–2454.
Zhou YF, Abbott RJ, Jiang ZY, Du FK, Milne RI, Liu JQ (2010). Gene flow and species delimitation: a case study of two pine species with overlapping distributions in southeast China. Evolution 64–8: 2342–2352.
Acknowledgements
We thank Master Hongtao Zhu for help in collecting needles and seeds for this study. We also thank Dr Zhonghu Li, Yongshuai Sun, Master Lili Wu and Quanjun Hu for their help in analyzing data. The paper benefited greatly from the comments received from four anonymous reviewers. The research was supported by grants from the National Natural Science Foundation of China (30725004) and Key Innovation Project of Ministry of Education of China to JQL, and a Royal Society-NSF China International Joint Project award 2010/R4 to RJA and JQL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Ren, GP., Abbott, R., Zhou, YF. et al. Genetic divergence, range expansion and possible homoploid hybrid speciation among pine species in Northeast China. Heredity 108, 552–562 (2012). https://doi.org/10.1038/hdy.2011.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.123
Keywords
This article is cited by
-
Analysis of the chloroplast genomes of four Pinus species in Northeast China: Insights into hybrid speciation and identification of DNA molecular markers
Journal of Forestry Research (2022)
-
Cytoplasmic DNA variation does not support a recent contribution of Pinus sylvestris L. from the Caucasus to the main range
Tree Genetics & Genomes (2020)
-
Genetic analysis of admixture and hybrid patterns of Populus hopeiensis and P. tomentosa
Scientific Reports (2019)
-
Importance of incomplete lineage sorting and introgression in the origin of shared genetic variation between two closely related pines with overlapping distributions
Heredity (2017)
-
Phylogeny and biogeography of Primula sect. Armerina: implications for plant evolution under climate change and the uplift of the Qinghai-Tibet Plateau
BMC Evolutionary Biology (2015)
