
Abstract
Local adaptation is considered a paradigm in studies of salmonid fish populations. Yet, little is known about the geographical scale of local adaptation. Is adaptive divergence primarily evident at the scale of regions or individual populations? Also, many salmonid populations are subject to spawning intrusion by farmed conspecifics that experience selection regimes fundamentally different from wild populations. This prompts the question if adaptive differences between wild populations and hatchery strains are more pronounced than between different wild populations? We addressed these issues by analyzing variation at 74 microsatellite loci (including anonymous and expressed sequence tag- and quantitative trait locus-linked markers) in 15 anadromous wild brown trout (Salmo trutta L.) populations, representing five geographical regions, along with two lake populations and two hatchery strains used for stocking some of the populations. FST-based outlier tests revealed more outlier loci between different geographical regions separated by 522±228 km (mean±s.d.) than between populations within regions separated by 117±79 km (mean±s.d.). A significant association between geographical distance and number of outliers between regions was evident. There was no evidence for more outliers in comparisons involving hatchery trout, but the loci under putative selection generally were not the same as those found to be outliers between wild populations. Our study supports the notion of local adaption being increasingly important at the scale of regions as compared with individual populations, and suggests that loci involved in adaptation to captive environments are not necessarily the same as those involved in adaptive divergence among wild populations.
Similar content being viewed by others
Introduction
When populations within a species experience different environmental conditions, the local selection regimes may eventually result in local adaptation, that is, populations exhibit higher fitness in their native environments than non-native populations translocated into the same environments (the ‘local versus foreign’ criterion as defined by Kawecki and Ebert, 2004). The presence and extent of local adaptation is thus a key issue both in general evolutionary biology and in conservation biology. In the latter context, evidence of local adaptation weighs heavily in the designation of conservation units (Fraser and Bernatchez, 2001). Moreover, considerations about preserving local adaptations within populations should be—but are unfortunately not always—essential elements in decision making about supplementing local populations by releasing non-local individuals (Tallmon et al., 2004).
Salmonid fishes are renowned for their tendency to form local, genetically differentiated populations, mediated in part by their well-developed homing instinct (Taylor, 1991). This has led to the general notion that salmonid populations are locally adapted. There is a considerable body of mostly circumstantial evidence suggesting local adaptation in many species of salmonid fishes (Taylor, 1991; Garcia de Leaniz et al., 2007; Fraser et al., 2011). However, relatively few studies would qualify as full documentation for having demonstrated local adaptation, for example, by demonstrating that the traits studied have a genetic basis and that differences among populations at the phenotypic or genic levels reflect selection as opposed to drift (Endler, 1986).
Recently, the development of statistical and conceptual frameworks such as the QST–FST approach (Merilä and Crnokrak, 2001) has enabled more rigorous testing of selection and local adaptation at the phenotypic level among salmonid populations (Koskinen et al., 2002; Perry et al., 2005; Jensen et al., 2008). Moreover, the revolutionizing developments in the genomic sciences have led to the generation of vast amounts of molecular resources available in silico, which can readily be used for identification of suitable genetic markers (Bouck and Vision, 2007). Combined with novel statistical approaches for detecting loci subject to (hitch-hiking) selection (Storz, 2005), this allows for addressing questions about local adaptation at the DNA level in salmonid fishes (Vasemägi et al., 2005; Aguilar and Garza, 2006; Hansen et al., 2010; Tonteri et al., 2010). Studies applying this type of genome scan approach have now been undertaken in a variety of organisms and have provided information about the proportion of the genomes involved in adaptive divergence, the candidate loci underlying adaptation and, by combining genome scans and quantitative genetics approaches, the genetic architecture of the specific phenotypic traits that are subject to selection (for example, Vasemägi et al., 2005; Rogers and Bernatchez, 2007; Kane and Rieseberg, 2007; Poncet et al., 2010). Compared with quantitative genetics approaches that require rearing of populations in common garden set-ups, analysis of footprints of selection is logistically simpler and thereby provides a realistic alternative for studies of local adaptation encompassing large geographical scales and many populations.
Although it is likely that many salmonid populations are locally adapted, it is not necessarily the case that all populations are locally adapted. In particular, information is mostly lacking concerning the geographical scale at which local adaptation takes place. From a theoretical viewpoint and assuming a stepping-stone model of gene flow, this question can be targeted by considering the relative roles of gene flow, drift and selection along with the geographical scale at which selection regimes are similar (Adkison, 1995); for instance, neighboring rivers can experience different selection regimes or several neighboring rivers within a region can experience similar selection regimes. Hansen et al. (2002) used estimated values of effective population sizes and rates of gene flow combined with hypothetical estimates of selection coefficients to evaluate the potential for local adaptation in anadromous brown trout (Salmo trutta) populations in Denmark. They concluded that local adaptation was more likely to occur on a regional basis encompassing several neighboring rivers rather than on the basis of individual population, unless selection was very strong (s>0.1). Similar conclusions have been reached for Pacific salmonids assuming realistic demographic parameters (Adkison, 1995), although it should be noted that Vähä et al. (2008) used the same approach in a study of tributary populations of Atlantic salmon (Salmo salar) and concluded that local adaptation was likely to occur at smaller geographical scales within a river system. Dionne et al. (2008) analyzed Atlantic salmon populations using landscape genetics methods and observed a hierarchical genetic structure that coincided with environmental variables, thereby also providing indirect evidence for local adaptation primarily occurring at the scale of regions rather than at the level of individual populations. Finally, a review of studies investigating local adaptation in salmonids at different geographical scales by directly estimating fitness showed that adaptation generally becomes more prevalent as geographical distance increases (Fraser et al., 2011). There were nevertheless also examples of local adaption at the scale of a few kilometers. Hence, the geographical scale at which local adaptation occurs in salmonid fishes remains unclear, and only few empirical studies have directly targeted this question.
A related issue concerns the relative importance of local adaptation among wild salmonid populations in comparison with adaptation to wild versus captive environments. Many salmonid species are subject to deliberate stocking or accidental escapes by farmed strains that have often been reared in captivity for many decades (Hindar et al., 1991; Hutchings and Fraser, 2008). The hatchery environments differ substantially from the conditions experienced by wild populations. For instance, reproduction is conducted by stripping parent fish, temperature regimes are regulated to improve growth rates, the fish are fed and no predators are present (Hindar et al., 1991). Furthermore, at least for brown trout the whole life cycles takes place under freshwater conditions although the strains may have been founded from anadromous populations (Hansen et al., 2010). This is likely to lead to inadvertent domestication selection, and many commercial strains are furthermore subject to deliberate selection programs. Several studies have documented fitness loss of farmed strains in natural environments as compared with wild populations (Hansen, 2002; McGinnity et al., 2003; Araki et al., 2007). In a recent study, Hansen et al. (2010) analyzed potential selection at 60 microsatellite loci in three wild brown trout populations and two hatchery strains used for stocking them. There was limited evidence for selection among the wild populations, but there was evidence for three loci being under diversifying selection between wild and hatchery strain trout. This could suggest that adaptation to wild versus captive environments is more pronounced than local adaptation among wild populations. However, further test of this hypothesis would require analysis of a larger number of populations in order to provide a better quantification and comparison of the numbers of loci under putative diversifying selection among wild populations and between captive and wild populations.
In this study, we took advantage of novel genome scan approaches for testing hypotheses about the scale of local adaptation that would be logistically unfeasible to pursue using quantitative genetics experiments. We analyzed footprints of selection among wild and hatchery populations of brown trout, based on 74 microsatellite loci. The samples encompassed 15 anadromous populations, two lake populations (see Figure 1) and two hatchery strains used for stocking Danish brown trout populations. Using outlier tests for identifying possible diversifying selection (Excoffier et al., 2009) and landscape genomics approaches for associating specific loci with environmental conditions (Joost et al., 2007), we tested the following hypotheses: (1) footprints of selection are more evident among regions separated by 522±228 km (mean±s.d.) than between populations within regions separated by 117±79 km (mean±s.d.). (2) Footprints of selection are more pronounced among hatchery versus wild populations than among wild populations.

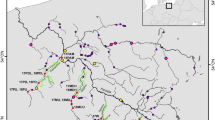
Map showing the location of sampled populations, sampling year and sample size. The different colors represents the different sampled regions;  Western Jutland (Wild);
Western Jutland (Wild);  Western Jutland (significantly admixed with hatchery trout);
Western Jutland (significantly admixed with hatchery trout);  Hadanger Fjord;
Hadanger Fjord;  Limfjord;
Limfjord;  East Jutland;
East Jutland;  Bornholm;
Bornholm;  Lake. Har-GRA, Granvin River; Har-GUD, Guddal River; WestW-STO, Storaa River; WestH-SKJ, Skjern River; WestH-VAR, Varde River; WestH-SNE, Sneum River; WestW-KON, Kongeaa River; WestW-RIB, Ribe River; Lim-KAR, Karup River; Lim-SKA, Skals River; East-VIL, Villestrup River; East-LIL, Lilleaa River; East-KOL, Kolding River; Born-TEJ, Tejn River; Born-LAE, Laesaa River; Lake-HAL, Lake Hald; Lake-MOS, Lake Mossoe; Hatch-VOR, Vork hatchery strain; Hatch-HK, Haarkaer Hatchery strain. A map of Europe showing the region studied is depicted in the upper right corner.
Lake. Har-GRA, Granvin River; Har-GUD, Guddal River; WestW-STO, Storaa River; WestH-SKJ, Skjern River; WestH-VAR, Varde River; WestH-SNE, Sneum River; WestW-KON, Kongeaa River; WestW-RIB, Ribe River; Lim-KAR, Karup River; Lim-SKA, Skals River; East-VIL, Villestrup River; East-LIL, Lilleaa River; East-KOL, Kolding River; Born-TEJ, Tejn River; Born-LAE, Laesaa River; Lake-HAL, Lake Hald; Lake-MOS, Lake Mossoe; Hatch-VOR, Vork hatchery strain; Hatch-HK, Haarkaer Hatchery strain. A map of Europe showing the region studied is depicted in the upper right corner.
Materials and methods
Sampled populations
We analyzed 19 brown trout populations from seven geographical and/or environmental groupings, delimited by the North Sea coast in the west to the Baltic Sea in the east and the Hardanger Fjord in Norway to the north (Figure 1). Specifically, this encompassed six populations from rivers on the North Sea Coast, western Jutland, two populations from the Limfjord region, Jutland, three populations from rivers on the east coast of Jutland, two populations from Bornholm Island in the Baltic Sea, two populations from the Hardanger Fjord, two lake-dwelling populations from Jutland and, finally, two hatchery strains that had been used for stocking the trout populations in western Jutland. These two hatchery strains have been founded by trout from eastern Jutland before at least the 1960s, and subsequent exchange of fish between the strains is known to have taken place (Hansen et al., 2001). A previous study showed extensive admixture (between 31 and 64%) by these hatchery strains in three of the western Jutland populations (Hansen et al., 2009), and they were therefore analyzed as a separate group.
The groupings of populations were defined primarily based on their geographical locations. Hence, there are natural geographical boundaries separating all the marine regions from each other. For instance, the populations in the Limfjord are separated from the North Sea and the east coast of Jutland by narrow straits, although obviously this does not preclude some gene flow among anadromous populations. Moreover, significant isolation-by-distance has been observed among brown trout populations within regions (Hansen and Mensberg, 1998; Hansen et al., 2009) demonstrating closer genetic relationships among populations within as opposed to among regional groups. Two of the groups, lake-dwelling and hatchery strain trout were defined based on environmental conditions rather than geographical proximity. In the case of hatchery trout, the grouping is, however, further justified by close genetic relationships and a common history of the strains (Hansen et al., 2001), and the two lake populations belong to the same river system (the Gudenaa River).
Samples consisted of adipose fin clips from electrofished trout caught between 1997 and 2009, with the exception of the Laesaa River sample, which consisted of scales collected using a smolt trap. Sample years and sample sizes are given in Figure 1. Population and region abbreviations are listed in Table 1.
Molecular analyses
DNA was extracted from samples using the DNeasy kit (QIAGEN, Hilden, Germany). We analyzed 74 microsatellite loci. These included 52 anonymous microsatellite markers along with 22 markers that have shown to be either expressed sequence tag (EST) or quantitative trait locus (QTL) linked in other salmonid species. In all, 54 of the markers have been mapped in brown trout representing 34 different linkage groups (Gharbi et al., 2006). For a full list of loci, including linkage relationships and possible functional relationships (QTLs or expressed sequence tags) we refer to Supplementary Tables S1 and S2, respectively.
QIAGEN's Multiplex PCR Kit was used for PCR amplification according to the manufacturer's recommendations, using an annealing temperature of 57 °C. Four loci with different terminal dyes were amplified in each multiplex. Several different combinations of loci were used in the multiplex sets (further information is available from the investigators on request). The loci were analyzed on an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's recommendations.
General statistical analyses
Exact test for deviations from Hardy–Weinberg equilibrium and linkage disequilibrium were analyzed with GENEPOP 4.0.7 (Raymond and Rousset, 1995). Observed (HO) and expected (HE) heterozygosity and allelic richness was quantified with FSTAT 2.9.1 (Goudet, 1995). Genetic differentiation between populations was estimated with θST (Weir and Cockerham, 1984) and 95% confidence intervals were determined by bootstrapping 10 000 times over loci using MSA 4.05 (Dieringer and Schlötterer, 2003). A hierarchical analysis of molecular variance was used for quantifying genetic differentiation among populations within and between regions, as implemented in the software Arlequin 3.5 (Excoffier et al., 2005). All tests for significance of pair-wise θST, Hardy–Weinberg equilibrium and linkage disequilibrium were adjusted for multiple tests by false discovery rate (FDR) correction (Benjamini and Yekutieli, 2001).
The software BOTTLENECK 1.2.02 (Cornuet and Luikart, 1996) was used to test for recent drastic population declines. The analyses were based on 10 000 iterations and assumed a two phase model with 90% stepwise and 10% non-stepwise mutations.
Outlier tests for selection
In order to detect loci that were possibly under selection, we applied a FST-based outlier test. This method is a modified version of Beaumont and Nichols (1996) method that is able to take hierarchical population structure into account (Excoffier et al., 2009). It is implemented in the software ARLEQUIN 3.5 (Excoffier et al., 2005). Briefly, the method detects outliers exhibiting significantly high or low FST values, controlled for heterozygosity at the loci. Outliers are detected based on differentiation among all populations (FST) and differentiation among groups of populations (FCT). The method assumes a hierarchical Island Model and simulates a large number of loci with different expected heterozygosity, which are used for generating confidence limits for the FST and FCT values. In cases of no hierarchical structure, the method becomes identical to that of Beaumont and Nichols (1996). As our study aimed at detecting adaptive divergence ultimately reflecting local adaptation, we focused on outliers suggested to be under diversifying selection. Although the method also allows for detecting outliers showing significantly low FST indicating balancing selection, this fell outside the topic of this study. Moreover, interpretation of results suggesting balancing selection may be complicated by mutation rate causing highly mutating loci to become outliers in terms of significantly low FST (Beaumont, 2008).
The analyses were conducted at three levels. (1) An overall analysis encompassing all analyzed populations (based on FST and FCT). (2) An analysis at the regional level conducting tests for pair-wise comparisons between analyzed regions (FCT). (3) Tests for outliers between populations within regions (FST). We assumed a model of 25 groups each consisting of 50 populations and based the tests on 50 000 simulated loci for the full data set. For the pair-wise comparisons, we assumed a model of five groups and for the tests among populations within groups we only assumed 50 populations. By conducting pair-wise hierarchical tests we were able to assess if (1) loci were predominantly outliers at a regional scale (FCT) or among populations within single regions (FST) and (2) if loci generally tended to be outliers between hatchery strains and groups consisting of wild populations.
In order to further assess possible correlations between geographical distance and local adaptation, we conducted an ‘outlier-by-distance’ analysis at the scale of regions (the regions Hatch and WestH were omitted). The analysis was based on the number of outlier loci (in terms of FCT) and geographical distance between pairs of regions, the latter calculated as the mean geographical distance between populations from different regions. Pair-wise geographic distances were determined by approximating the shortest waterway distance between river mouths or lake outlets using Google Earth. We plotted numbers of outlier loci against geographical distance and further tested for association using a Mantel test implemented in IBDWS (Jensen et al., 2005).
Landscape genomics
In order to associate loci under possible selection with specific selection regimes, we tested for correlation between environmental variation and genetic variation at the specific loci. Factors such as temperature may vary considerably among and within rivers at smaller geographical scales and may represent potent selection regimes (Jensen et al., 2008). However, information at this geographical scale was not available for all sampled localities. We therefore concentrated on parameters associated with the marine environment encountered by anadromous trout leaving their natal river. These parameters are known to exhibit considerable variation on large geographical scales, such as salinity decreasing from the North Sea toward the Baltic Sea and temperature decreasing from south to north. The following two environmental factors were thus included: (1) salinity and (2) temperature. The specific data for salinity and temperature and their sources are listed in Table 1. Since information for the year 1997 was available for all populations, data from this period were used. The maximum records for both parameters at a depth of 1±0.5 m (total range) were used as these are expected to have the strongest selective impact.
We applied the spatial analysis method by Joost et al. (2007). The method tests for association between all possible pairs of environmental variables and allele frequencies. Univariate logistic regressions are used to determine the markers involved in the most significant models of the possible pairs. The significance of the regression coefficients are subsequently evaluated by tests comparing a model including the environmental variable and a model with a constant only. A Wald test implemented in the software was used to determine the significance of the models. We used the spatial analysis method test for identifying loci and alleles associated with salinity and temperature for all wild populations, but excluded WestH-SKJ, WestH-VAR and WestH-SNE because of strong admixture with stocked hatchery trout, and Lake-HAL and Lake-MOS because of missing environmental data for these populations.
Results
Genetic variation
Summary statistics for the microsatellite loci are listed in Supplementary Table S3. Number of alleles ranged from 2 at Ssa14DU to 70 at Str11INRA. Significant deviations from Hardy–Weinberg equilibrium were observed in 198 of a total of 1406 tests after FDR correction. Five loci (SSOSL32, Ssa161NVH, Ssa4DIAS, UBA and Ssa156NVH) accounted for 66 of these deviations, showing heterozygote deficits suggestive of null alleles. Tests for linkage disequilibrium revealed significant associations between UBA and TAP2B in six of the analyzed populations after FDR correction. These two loci are known to be physically linked (Grimholt et al., 2002). Ssa408UoS and Ssa161NVH showed significant association in four populations and these two loci both map to linkage group 13 (Gharbi et al., 2006). No clear patterns were revealed for the remaining linkage groups, indicating that the markers were either unlinked or weakly physically linked. In this study, we therefore refer to loci rather than linkage groups as being under possible selection, but we stress that hitch-hiking selection rather than selection at the specific loci is likely to occur.
Pair-wise θST values ranged from 0.003 between WestW-KON and WestW-RIB to 0.132 between Har-GRA and Hatch-HK (Table 2). A hierarchical analysis of molecular variance showed significant genetic differentiation both among samples within groups (FSC=0.027, P<0.001) and at the between-group level (FCT=0.023, P<0.001). Genetic differentiation, FST among all samples and based on all markers was 0.049 (P<0.001). The tests for recent bottlenecks yielded no significant outcomes in any of the populations.
Outlier tests for selection
The hierarchical outlier test by Excoffier et al. (2009) suggested two loci to be under diversifying selection across all populations (FST; SSOSL438 and Ssa19NVH) (Figure 2a), whereas five outliers were detected between regions (FCT; Ssa14DU, Str73, Ssa85, Ssa55NVH and Ssa207NVH) (Figure 2b). However, this global analysis encompassing populations from a range of potentially different selection regimes may mask footprints of selection between specific contrasting selection regimes. We therefore conducted pair-wise tests at the regional level (that is, detected outliers at the between-region level, FCT) and within regions between populations (FST). By contrasting the results for outlier tests between regions (FCT) with outlier tests involving populations within each region (FST) it was possible to assess if selection was more pronounced between regions or among populations within regions.

Results of the hierarchical outlier test involving all loci and all samples. (a) Within regions: FST values are plotted against heterozygosity. (b) Between regions: FCT values are plotted against heterozygosity. Long dashed lines denote 0.01 and 0.99 quantiles, short dashed lines denote 0.05 and 0.95 quantiles. Loci with open circles are candidates for being under diversifying selection.
Pair-wise tests between regions revealed a high number of outliers potentially under selection (Table 3). The highest numbers of outliers were found in comparisons involving either Har, Born, Lake or Hatch regions. These regions are those deviating the most in terms of environmental conditions, that is, salinity, temperature and captive environments, but in the case of Har and Born these regions are also the geographically most remote. The aspect of geographical distance was further investigated by the ‘outlier-by-distance’ analysis, which showed a significant positive correlation between geographical distance and the number of outlier loci between regions (r2=0.43, P=0.035; Figure 3a). However, as this likely included several false positives we repeated the analysis by including only loci that were found to be significant outliers after correcting for the FDR. This yielded a lower, but still significant correlation between numbers of outliers versus geographical distance (r2=0.27, P=0.020; Figure 3b).

Numbers of outlier loci between pairs of regions plotted against geographical distance. Outlier loci were identified by a hierarchical outlier test (Excoffier et al., 2009) based on FCT between regions. The regions Hatch and WestH were omitted. (a) All outliers detected between regions. (b) Significant outliers after FDR correction.
There was no clear tendency toward more loci being outliers in hatchery–wild than wild–wild comparisons. Focusing on regions around the Jutland Peninsula, three outliers were observed in WestW versus Hatch and East versus Hatch, but seven outliers between Lim versus Hatch and Lake versus Hatch (Table 3).
Some loci were outliers in several different comparisons; Oneμ2, Ssa55NVH, Ssa156NVH, Str73, OmyRGT14TUF and Str58CNRS were outliers in eight or seven pair-wise comparisons (Tables 3 and 4). OmyRGT14TUF, Ssa207NVH, Ssa94NVH, Ssa14DU and SSOSL417 were outliers in several comparisons involving the populations from the Har region (Table 3). Oneμ2, Ssa55NVH and MST-543 were outliers in comparisons involving the Born region. Str73 was an outlier in comparisons involving the lake region, and Ssa156NVH was an outlier in comparisons involving both Born and lake regions. Finally, CA060208, Ssa103NVH, Str58CNRS and Ssa119NVH were outliers in several comparisons involving hatch (Table 3).
In contrast to the outlier tests between regions, the tests involving populations within regions yielded considerably fewer outliers (Table 3). Few of the loci found consistently to be outliers between regions were also outliers among populations within regions. A χ2-test further showed significant differences in proportion of outliers between as opposed to within regions (χ2=7.508, df=1, P=0.006).
Landscape genomics
The spatial analysis method (Joost et al., 2007) revealed several outliers after Bonferroni correction. As there is limited information in alleles that are rare or close to fixation, we removed alleles with frequencies above 95 and below 5% in the total data set according to Joost et al. (2007). Out of 379 alleles, 46 were found to be significantly correlated with maximum salinity and 25 with maximum temperature (at the 1% level). At a 0.1% level, 30 alleles were found to be significantly correlated with salinity and 14 alleles with temperature. The results for the spatial analysis method analysis are listed in Table 4 for those loci, which were also found to be outliers between regions using hierarchical outlier tests.
Discussion
Our study provided evidence that diversifying selection is more pronounced between regions compared with populations within regions and that geographical scale has an important role in the prevalence of diversifying selection and thereby local adaptation in brown trout populations. There was no clear tendency toward footprints of selection being more pronounced between hatchery trout and wild trout than between wild trout from different populations. However, the analyses pointed toward specific loci being subject to diversifying selection in captive environments that were not to the same extent outliers between wild populations. We discuss these points below along with a general evaluation of the evidence for selection.
Evidence for selection
Several outlier loci possibly under diversifying selection were detected. This is clearly not to say that all of these loci are under diversifying selection; indeed, there are likely to be several false positives among them. Severe bottlenecks represent one source of error that may cause loci to be erroneously identified as being under selection (Teshima et al., 2006). However, none of the tests for recent bottlenecks generated significant outcomes, despite strong statistical power given the high number of loci. Multiple testing is another important factor resulting in false positives. We applied a FDR correction when appropriate, but for the many FST-based tests by Beaumont and Nichols (1996) and Excoffier et al. (2009) this procedure would be very conservative (see Table 3). Thus, emphasis was also put on the number of times a locus was found to be an outlier in different comparisons.
Most importantly, however, we stress that a major purpose of this study was to compare the number of outliers between populations within regions versus number of outliers occurring between regions. We note that violations of the model implemented in the hierarchical outlier method (that is, using the wrong genetic structure) can result in a higher number of false positives. However, our comparison of regional versus local levels are relative and should not be strongly affected by the presence of false positives, unless there is a bias in their occurrence at different geographical levels, or alternatively different statistical power for tests at different levels. We detected outliers among populations within regions using Beaumont and Nichols (1996) test, based on FST, and outliers between regions using the related hierarchical test by Excoffier et al. (2009), based on FCT. We cannot rule out that statistical power of tests differs for these two estimators. Nevertheless, we find that our conclusion that footprints of selection are more evident at regional scales rather than local scales appear robust. First, it is evident from Tables 3 and 4 that several loci were outliers in several (up to eight) different tests among regions, strengthening their candidacy for being under diversifying selection. In contrast, no single locus was an outlier in more than two tests within regions. Second, the significant association between numbers of outliers and geographical distance between regions provide further evidence for the importance of geographical scale in local adaptation. Finally, a χ2-test revealed that the proportion of outliers between regions was significantly different from the proportion of outliers within regions.
The spatial scale of local adaptation
The majority of studies so far of local adaptation in salmonid fishes have aimed generally at detecting adaptation per se (reviewed in Taylor, 1991; Garcia de Leaniz et al., 2007 and Fraser et al., 2011). This is not surprising, given the formidable workload and logistic problems associated with studies measuring fitness or fitness-related traits; studies systematically analyzing populations across geographical regions would in most cases be unfeasible, except for reviews and analyses of published studies (Fraser et al., 2011). However, our study shows that population genomics approaches can be used for resolving this issue. In their seminal study of footprints of selection in Atlantic salmon populations, Vasemägi et al. (2005) employed a hierarchical sampling design in order to identify loci under selection at local and regional scales, but did not provide a further biological interpretation of the results. In this study, we systematically compared evidence for selection at different geographical scales and observed more outlier loci between regions separated by 522±228 km (mean±s.d.) than among populations within regions separated by 117±79 km (mean±s.d.). Also a significant positive association between geographical distance and numbers of outliers between regions was observed and the proportions of outliers between regions compared with within regions were significantly different. Hence, these findings suggest an important link between geographical distance and diversifying selection, and thereby implicitly local adaptation.
Whether or not geographical distance per se is important or confounded by environmental variation across large distance is debatable. The studied localities covered a wide range of habitats and environmental conditions. For instance, considering the marine environments that anadromous trout would encounter, these ranged from brackish in the Baltic Sea around Bornholm Island to oceanic salinities in the North Sea, which western Jutland rivers flow into. A study of single-nucleotide polymorphism variation in Atlantic cod (Gadus morhua) across these regions revealed several outlier loci correlated with salinity and temperature (Nielsen et al., 2009). The landscape genomics analyses of this study revealed a similar pattern. For several of the outlier loci detected by the FST-based outlier test, one or more alleles were significantly associated with temperature, salinity or both, suggesting a link between outlier status of these loci and environmental variation across localities.
In total, and in accordance with the review by Fraser et al. (2011), we argue that the prevalence of local adaptation increases with geographical distance. However, we also note that this is likely to reflect increasingly different selection regimes, and within smaller regions with highly heterogenous selection regimes the conclusion might be different. Finally, it should be considered that different loci may respond to different selection regimes. For instance, pathogens and parasites may show patchy distribution at microgeographical scales (Bakke and Harris, 1998). Correspondingly, selection at microgeographical scales has been observed at immune-related loci in salmonid fishes (Landry and Bernatchez, 2001; Miller et al., 2001; Aguilar and Garza, 2006). At the other end of the scale, CLOCK genes are involved in photoperiodicity and determination of seasonal timing. They would therefore be expected to respond to selection regimes on much larger geographical scales, which has also been observed in Chinook salmon (Oncorhynchus tshawytscha) populations along thousands of kilometers of the North American Pacific coast (O'Mally and Banks, 2008). Although certain general patterns of relationships between local adaptation and geographical distance may be expected, it is therefore nevertheless also evident that variation exists among loci depending on their functional role.
Diversifying selection in hatchery versus wild trout
We did not find general support for the hypothesis that diversifying selection should be more prevalent in hatchery versus wild trout as compared with diversifying selection among wild trout populations. However, there was considerable consistency in the loci being outliers in hatchery–wild comparisons, primarily Str58CNRS, CA060208, Ssa119NVH and Ssa103NVH (Table 3). Except for Str58CNRS, these loci were only in a couple of cases outliers between wild populations and regions. This suggests that diversifying selection between wild and hatchery strain trout involves different loci than those under selection among wild populations. Although this latter point is based on analysis of relatively few loci, it does receive support from an analysis of transcriptome differences between farmed and wild Atlantic salmon (Roberge et al., 2006). Here, farmed and wild salmon showed considerable divergence, but different farmed strains of European and North American origin showed convergence at the transcriptome level, indicating parallel evolution during the domestication process.
Hansen et al. (2010) analyzed a subset of the same loci in the two hatchery strains and historical (1943–1956) and contemporary samples from three of the western Jutland populations also analyzed in this study (WestH-SKJ, WestW-KON and WestW-STO). This spatiotemporal analysis suggested three loci as candidates for being under diversifying selection in hatchery versus wild trout; CA060208, Ssa19NVH and Ssa63NVH. On the larger geographical scale of this study, CA060208 still emerged as a candidate locus. Ssa19NVH was also suggested to be under diversifying selection but not in hatchery–wild trout comparisons, whereas Ssa63NVH was only an outlier in a single case (Table 3).
Possible functional relationship of detected outliers
Several of the outliers detected in this study have been suggested to be linked to functional genes. This particularly concerned SSOSL32, Ssa85 and Ssa14DU (see Supplementary Table S2 for details). We noted with particular interest that the four loci consistently showing outlier status in comparisons between wild and hatchery populations had previously been suggested to be linked to functional genes in other salmonids. Ssa103NVH is linked to a spawning time QTL in rainbow trout (Oncorhynchus mykiss) (O'Malley et al., 2003), Str58CNRS is linked to a QTL for body weight in Atlantic salmon (Reid et al., 2005), Ssa119NVH is linked to a QTL for upper temperature tolerance in rainbow trout (Jackson et al., 1998) and Arctic charr (Salvelinus alpinus) (Somorjai et al., 2003), and CA060208 is found in an expressed sequence tag of a gene with unknown function, but suggested to be a candidate locus for freshwater versus saltwater adaptation in Atlantic salmon (Vasemägi et al., 2005). It would make sense if inadvertent selection for spawning time, body weight, temperature tolerance and adaptation to a constant freshwater environment had occurred in hatchery strain trout. The landscape genomics analysis (Joost et al., 2007) also suggested associations between an allele at Ssa103NVH and temperature in the marine environment. Similarly, for Str58CNRS one allele was associated with salinity (Table 4). These results suggest that the two loci found to be QTLs in other salmonids may also be linked to ecologically important functional variation in brown trout.
Conclusions
This study provides one of the first empirical assessments of the spatial scale of local adaptation at the molecular genetic level in salmonid fishes. Along with other studies, the results illustrate that the dynamics of local adaptation cannot be understood solely at the level of individual populations, but integrate geographical distance, spatially varying environmental conditions and demographic parameters such as gene flow (Adkison, 1995; Hansen et al., 2002; Dionne et al., 2008; Fraser et al., 2011). Our findings also suggest that diversifying selection between hatchery and wild salmonids may involve different loci than those under selection between wild populations. This study was based on microsatellite analysis. However, with the ever increasing development of next generation sequencing and single-nucleotide polymorphism genotyping, we foresee a drastic increase in our knowledge about local adaption both in terms of its geographical extent and the underlying loci and pathways (for example, Nielsen et al., 2009; Hohenlohe et al. 2010; Renaut et al., 2010).
References
Adkison MD (1995). Population differentiation in Pacific salmon: local adaptation, genetic drift, or the environment? Can J Fish Aquat Sci 52: 2762–2777.
Aguilar A, Garza JC (2006). A comparison of variability and population structure for major histocompatibility complex and microsatellite loci in California coastal steelhead (Oncorhynchus mykiss Walbaum). Mol Ecol 15: 923–937.
Araki H, Cooper B, Blouin MS (2007). Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science 318: 100–103.
Bakke TA, Harris PD (1998). Diseases and parasites in wild Atlantic salmon (Salmo salar) populations. Can J Fish Aquat Sci 55: 247–266.
Beaumont MA (2008). Selection and sticklebacks. Mol Ecol 17: 3425–3427.
Beaumont MA, Nichols RA (1996). Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B Biol Sci 263: 1619–1626.
Benjamini Y, Yekutieli D (2001). The control of the false discovery rate in multiple testing under dependency. Ann Stat 29: 1165–1188.
Bouck A, Vision T (2007). The molecular ecologist's guide to expressed sequence tags. Mol Ecol 16: 907–924.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Dieringer D, Schlötterer C (2003). Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3: 167–169.
Dionne M, Caron F, Dodson JJ, Bernatchez L (2008). Landscape genetics and hierarchical genetic structure in Atlantic salmon: the interaction of gene flow and local adaptation. Mol Ecol 17: 2382–2396.
Endler JA (1986). Natural Selection in the Wild. Princeton University Press: Princeton.
Excoffier L, Hofer T, Foll M (2009). Detecting loci under selection in a hierarchically structured population. Heredity 103: 285–298.
Excoffier L, Laval G, Schneider S (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Fraser DJ, Weir LK, Bernatchez L, Hansen MM, Taylor EB (2011). Extent and scale of local adaptation in salmonid fishes: review and meta-analysis. Heredity 106: 404–420.
Fraser DJ, Bernatchez L (2001). Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10: 2741–2752.
Garcia de Leaniz C, Fleming IA, Einum S, Verspoor E, Jordan WC, Consuegra S et al. (2007). A critical review of adaptive genetic variation in Atlantic salmon: implications for conservation. Biol Rev 82: 173–211.
Gharbi K, Gautier A, Danzmann RG, Gharbi S, Sakamoto T, Høyheim B et al. (2006). A linkage map for brown trout (Salmo trutta): chromosome homeologies and comparative genome organization with other salmonid fish. Genetics 172: 2405–2419.
Goudet J (1995). FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Grimholt U, Drabløs F, Jørgensen SM, Høyheim B, Stet RJM (2002). The major histocompatibility class I locus in Atlantic salmon (Salmo salar L.): polymorphism, linkage analysis and protein modelling. Immunogenetics 54: 570–581.
Hansen MM (2002). Estimating the long-term effects of stocking domesticated trout into wild brown trout (Salmo trutta) populations: an approach using microsatellite DNA analysis of historical and contemporary samples. Mol Ecol 11: 1003–1015.
Hansen MM, Fraser DJ, Meier K, Mensberg KLD (2009). Sixty years of anthropogenic pressure: a spatio-temporal genetic analysis of brown trout populations subject to stocking and population declines. Mol Ecol 18: 2549–2562.
Hansen MM, Meier K, Mensberg KL (2010). Identifying footprints of selection in stocked brown trout populations: a spatio-temporal approach. Mol Ecol 19: 1787–1800.
Hansen MM, Mensberg KLD (1998). Genetic differentiation and relationship between genetic and geographical distance in Danish sea trout (Salmo trutta L.): populations. Heredity 81: 493–504.
Hansen MM, Ruzzante DE, Nielsen EE, Bekkevold D, Mensberg KLD (2002). Long-term effective population sizes, temporal stability of genetic composition and potential for local adaptation in anadromous brown trout (Salmo trutta) populations. Mol Ecol 11: 2523–2535.
Hansen MM, Ruzzante DE, Nielsen EE, Mensberg KLD (2001). Brown trout (Salmo trutta) stocking impact assessment using microsatellite DNA markers. Ecol Appl 11: 148–160.
Hindar K, Ryman N, Utter F (1991). Genetic effects of cultured fish on natural fish populations. Can J Fish Aquat Sci 48: 945–957.
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA (2010). Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. Plos Genet 6: e1000862.
Hutchings JA, Fraser DJ (2008). The nature of fisheries- and farming-induced evolution. Mol Ecol 17: 294–313.
Jackson TR, Ferguson MM, Danzmann RG, Fishback AG, Ihssen PE, O'Connell M et al. (1998). Identification of two QTL influencing upper temperature tolerance in three rainbow trout (Oncorhynchus mykiss) half-sib families. Heredity 80: 143–151.
Jensen JL, Bohonak AJ, Kelley ST (2005). Isolation by distance, web service. BMC Genet 6: 13.
Jensen LF, Hansen MM, Pertoldi C, Holdensgaard G, Mensberg KLD, Loeschcke V (2008). Local adaptation in brown trout early life-history traits: implications for climate change adaptability. Proc R Soc B Biol Sci 275: 2859–2868.
Joost S, Bonin A, Bruford MW, Despres L, Conord C, Erhardt G et al. (2007). A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16: 3955–3969.
Kane NC, Rieseberg LH (2007). Selective sweeps reveal candidate genes for adaptation to drought and salt tolerance in common sunflower, Helianthus annuus. Genetics 175: 1823–1834.
Kawecki TJ, Ebert D (2004). Conceptual issues in local adaptation. Ecol Lett 7: 1225–1241.
Koskinen MT, Haugen TO, Primmer CR (2002). Contemporary fisherian life-history evolution in small salmonid populations. Nature 419: 826–830.
Landry C, Bernatchez L (2001). Comparative analysis of population structure across environments and geographical scales at major histocompatibility complex and microsatellite loci in Atlantic salmon (Salmo salar). Mol Ecol 10: 2525–2539.
McGinnity P, Prodöhl P, Ferguson K, Hynes R, Maoiléidigh Nó, Baker N et al. (2003). Fitness reduction and potential extinction of wild populations of Atlantic salmon, Salmo salar, as a result of interactions with escaped farm salmon. Proc R Soc Lond B Biol Sci 270: 2443–2450.
Merilä J, Crnokrak P (2001). Comparison of genetic differentiation at marker loci and quantitative traits. J Evol Biol 14: 892–903.
Miller KM, Kaukinen KH, Beacham TD, Withler RE (2001). Geographic heterogeneity in natural selection on an MHC locus in sockeye salmon. Genetica 111: 237–257.
Nielsen EE, Hemmer-Hansen J, Poulsen NA, Loeschcke V, Moen T, Johansen T et al. (2009). Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua). BMC Evol Biol 9: 276.
O'Malley KG, Sakamoto T, Danzmann RG, Ferguson MM (2003). Quantitative trait loci for spawning date and body weight in rainbow trout: testing for conserved effects across ancestrally duplicated chromosomes. J Hered 94: 273–284.
O'Mally KG, Banks MA (2008). A latitudinal cline in the chinook salmon (Oncorhynchus tshawytscha) Clock gene: evidence for selection on PolyQ lenght variants. Proc R Soc B 275: 2813–2821.
Perry GML, Audet C, Bernatchez L (2005). Maternal genetic effects on adaptive divergence between anadromous and resident brook charr during early life history. J Evol Biol 18: 1348–1361.
Poncet BN, Herrmann D, Gugerli F, Taberlet P, Holderegger R, Gielly L et al. (2010). Tracking genes of ecological relevance using a genome scan in two independent regional population samples of Arabis alpina. Mol Ecol 19: 2896–2907.
Raymond M, Rousset F (1995). Genepop (version 1.2): a population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Reid DP, Szanto A, Glebe B, Danzmann RG, Ferguson MM (2005). QTL for body weight and condition factor in Atlantic salmon (Salmo salar): comparative analysis with rainbow trout (Oncorhynchus mykiss) and Arctic charr (Salvelinus alpinus). Heredity 94: 166–172.
Renaut S, Nolte AW, Bernatchez L (2010). Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae). Mol Ecol 19: 115–131.
Roberge C, Einum S, Guderley H, Bernatchez L (2006). Rapid parallel evolutionary changes of gene transcription profiles in farmed Atlantic salmon. Mol Ecol 15: 9–20.
Rogers SM, Bernatchez L (2007). The genetic architecture of ecological speciation and the association with signatures of selection in Natural Lake Whitefish (Coregonas sp. Salmonidae) species pairs. Mol Biol Evol 24: 1423–1438.
Somorjai IML, Danzmann RG, Ferguson MM (2003). Distribution of temperature tolerance quantitative trait loci in Arctic charr (Salvelinus alpinus) and inferred homologies in rainbow trout (Oncorhynchus mykiss). Genetics 165: 1443–1456.
Storz JF (2005). Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol Ecol 14: 671–688.
Tallmon DA, Luikart G, Waples RS (2004). The alluring simplicity and complex reality of genetic rescue. Trends Ecol Evol 19: 489–496.
Taylor EB (1991). A review of local adaptation in Salmonidae, with particular reference to Pacific and Atlantic Salmon. Aquaculture 98: 185–207.
Teshima KM, Coop G, Przeworski M (2006). How reliable are empirical genomic scans for selective sweeps? Genome Res 16: 702–712.
Tonteri A, Vasemägi A, Lumme J, Primmer CR (2010). Beyond MHC: signals of elevated selection pressure on Atlantic salmon (Salmo salar) immune-relevant loci. Mol Ecol 19: 1273–1282.
Vähä JP, Erkinaro J, Niemelä E, Primmer CR (2008). Temporally stable genetic structure and low migration in an Atlantic salmon population complex: implications for conservation and management. Evol Appl 1: 137–154.
Vasemägi A, Nilsson J, Primmer CR (2005). Expressed sequence tag-linked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in Atlantic Salmon (Salmo salar L.). Mol Biol Evol 22: 1067–1076.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Acknowledgements
We thank numerous anglers for sampling of trout, the Danish Natural Science Research Council (Grant no. 272-05-0202) for funding, Thomas Damm Als for computational assistance and Craig Primmer and two anonymous referees for numerous constructive comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Meier, K., Hansen, M., Bekkevold, D. et al. An assessment of the spatial scale of local adaptation in brown trout (Salmo trutta L.): footprints of selection at microsatellite DNA loci. Heredity 106, 488–499 (2011). https://doi.org/10.1038/hdy.2010.164
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.164
Keywords
This article is cited by
-
ddRAD-seq reveals the genetic structure and detects signals of selection in Italian brown trout
Genetics Selection Evolution (2022)
-
A landscape approach for identifying potential reestablishment sites for extirpated stream fishes: an example with Arctic grayling (Thymallus arcticus) in Michigan
Hydrobiologia (2022)
-
Failed protective effort of ex situ conservation of River Vistula trout (Salmo trutta) in Sweden
Environmental Biology of Fishes (2022)
-
Genetic and phenotypic variation along an ecological gradient in lake trout Salvelinus namaycush
BMC Evolutionary Biology (2016)
-
The population genomic signature of environmental selection in the widespread insect-pollinated tree species Frangula alnus at different geographical scales
Heredity (2015)
