
Abstract
Species that overlap over a large part of their range and habitat requirements are challenging for the study of speciation and hybridization. In this respect, the study of broadscale introgressive hybridization has raised recent interest. Here we studied hybridization between two closely related amphibians Lissotriton helveticus and Lissotriton vulgaris that reproduce over a wide sympatric zone. We used mitochondrial and microsatellite markers on 1272 individuals in 37 sites over Europe to detect hybrids at the individual-level and to analyse Hardy–Weinberg and linkage disequilibria at the population-level. Morphological traits showed a strong bimodal distribution. Consistently, hybrid frequency was low (1.7%). We found asymmetric introgression with five times more hybrids in L. vulgaris than in L. helveticus, a pattern probably explained by an unequal effective population size in a study part wherein L. helveticus numerically predominates. Strikingly, significant levels of introgression were detected in 73% of sites shared by both species. Our study showed that introgression is widespread but remains confined to the sites where the two species reproduce at the same time. This pattern may explain why these species remain genetically distinct over a broad sympatric zone.
Similar content being viewed by others

Introduction
Natural hybridization has long raised the interest of evolutionists as it represents a unique source of information about the mechanisms of reproductive isolation, which can lead to speciation (Coyne and Orr, 2004). In particular, the maintenance of species integrity in the face of gene flow remains a central outcome in evolutionary biology (Sites and Marshall, 2003; Hey, 2006; Baack and Rieseberg, 2007; Petit and Excoffier, 2009). The two main models proposed to describe the dynamics of the hybridization process, the Tension zone model and the Mosaic zone model, account for many situations observed in nature.
Barton and Hewitt (1985) have championed the concept of Tension zone in which clines are maintained by a balance between parental effective migration and selection against hybrids. The low fitness of hybrids is caused by the disruption of intragenomic co-adaption (that is, endogenous selection). The tension zone model performs well in situations of parapatry and when zone width is small relative to the breeding range of the two taxa (Endler, 1977). In contrast, the mosaic zone model (Harrison, 1986; Howard, 1986) assumes that two species segregate between two habitat types. Differential selection caused by ecological factors (that is, exogenous selection) maintains species integrity by limiting the possibility of successful dispersal into the other habitat.
Although the tension zone and the mosaic models describe a wide range of situations encountered in animals and plants, they are not universal and their assumptions are not always met (Harrison, 1993). Predictions about hybridization dynamics become less clear (i) when the zone of sympatry covers a large fraction of the breeding ranges of focal species, and (ii) when species are frequently found in syntopy. In other words, classical models are not suitable for cases wherein species exhibit broad geographical and ecological overlap. A plausible reason why these situations did not raise strong interest in previous studies is that hybridization is expected to occur at a very low rate between broadly sympatric taxa and that reproductive or other barriers are strong enough to maintain species integrity. Hybridization events can thus be viewed as having no biological significance in these cases. However, a broad overlap zone presents a large interface surface between two species so that even if hybridization events are locally rare, they are not necessarily so at the zone scale. In addition, exogenous selection against hybrids is expected to be negatively related to the degree of ecological overlap between species. The more the two species ecologically overlap, the less environmental components of fitness will favour parental forms over hybrids. In such situations, the mechanisms responsible for the maintenance of species integrity in mixed populations are of particular interest.
Several broad hybrid zones have been studied (Roques et al., 2001; Höbel and Gerhardt, 2003; Borge et al., 2005; Turner et al., 2005; Cooley et al., 2006; Duvernell et al., 2007; Fuller, 2008; Jang et al., 2009). However, in these studies attention was largely given to cases of invasion by an allochthonous or clonal species into the distribution of an indigenous/wild species (Rubidge and Taylor, 2004; Duputié et al., 2007) or a recent range expansion (Martin and Cruzan, 1999). Studies investigating cases of ancient contact between autochthonous taxa remain rare. Such studies would bring new insights to our understanding of the mechanisms involved in the maintenance of species integrity in setups that differ from the classical tension or mosaic zones.
We investigated hybridization and introgression between the palmate newt Lissotriton helveticus and the smooth newt Lissotriton vulgaris. These related taxa diverged approximately 44 million years ago (mya) (Steinfartz et al., 2007) to less than 20 mya (Weisrock et al., 2001). The last secondary contact probably formed after the last glaciation (Spurway and Callan, 1960). The zone of sympatry extends over a thousand kilometres in longitude and latitude in Europe (IUCN, 2008) and is broadly relative to the breeding range of each species (Figure 1) (72% of L. helveticus range and 11% of L. vulgaris range is sympatric). Despite both species having different ecological requirements (Denoël and Ficetola, 2007, 2008), they frequently breed in the same water bodies (Arntzen, 1986; Griffiths, 1987) where extensive overlap in microhabitat selection, diet and breeding period is observed (Griffiths, 1986, 1987; Chadwick et al., 2006). Heterospecific courtship is relatively common in the lab, which suggests that premating barriers are not strong (Wambreuse and Bels, 1984; Roberts, 1990; Secondi et al., 2010).

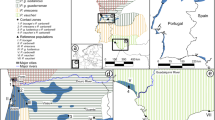
Sampled sites, distribution of Lissotriton helveticus and L. vulgaris and location of the zone of sympatry (from IUCN, 2008). The outlet shows the study site where we investigated patterns of introgression at a local scale (BD CARTHAGE IGN, Paris, France 2005). White circles indicate sampled sites where only L. helveticus were found and black triangles indicate sampled sites with only L. vulgaris. Grey rectangles indicate syntopic sites. Stars indicate the locations of hybrids as reported in the literature (a, Griffiths et al., 1987; b,c, Arntzen et al., 1998; d,e, Beebee et al., 1999; f, Schlüpmann et al., 1999).
Hybrids have been found in several locations (Griffiths et al., 1987; Arntzen et al., 1998; Beebee et al., 1999; Schlüpmann et al., 1999) (see Figure 1). Laboratory experiments suggested that if hybrids were less viable and fertile than the progeny of homospecific crosses (Spurway and Callan, 1960; Cogălniceanu, 1994), they could backcross and produce a viable offspring (Benazzi, 1957). An earlier study detected introgression in three areas of the sympatric zone (Arntzen et al., 1998). Only F1 hybrid males have been detected in wild adults so far (Griffiths et al., 1987; Arntzen et al., 1998; Beebee et al., 1999; Schlüpmann et al., 1999). The fact is striking as hybrid females are expected to be more viable than hybrid males (Haldane, 1922), males being the heterogametic sex in Lissotriton (Hillis and Green, 1990). Our current knowledge of the hybridization process between the two species may be strongly biased because of the difficulty in identifying phenotypic hybrids in one sex. In contrast to males that are readily identified by secondary sexual traits, females of each species look alike, and overlap for several traits such as colour pattern of underparts (Johanet et al., 2009b). Thus, female hybrids are difficult to identify and female backcrosses are almost impossible to distinguish from parental species (Naisbit et al., 2003) by morphological criteria alone. Moreover, in another study of hybridization between the small-bodied newts L. vulgaris and L. montandoni, even introgressed males were often indistinguishable from parental species (Babik et al., 2003).
By conducting a large scale study on hybridization between L. helveticus and L. vulgaris we addressed several questions that arise from two main objectives. First, we revaluated introgression and compared the efficiency of hybrid detection from morphometric and genotypic methods. We expected genotypic data to be more discriminant than morphometric ones. We also tested the effect of sex and species on introgression level. According to Haldane's rule (Haldane, 1922), we expected females to be more introgressed than males, as males are the heterogametic sex in Lissotriton (Hillis and Green, 1990). Second, we explored the maintenance of species integrity by comparison of introgression levels at different biogeographical scales. (i) We compared introgression levels inside the breeding ranges of the two species by sampling distant areas with different syntopic contexts (allopatry vs sympatry). We expected that limited introgression outside sympatry would maintain species integrity. (ii) To analyse introgression dynamics at the local scale (allotopy vs syntopy), we selected an area located at the southern edge of the sympatric zone. This area represented a unique configuration because L. vulgaris is always encountered syntopically with L. helveticus in floodplain valleys; the latter species is also found in allotopy in valley or plateau sites. The nesting of L. vulgaris habitat within L. helveticus habitat is an extreme case of ecological overlap between two hybridizing species. In this study area, the occurrence ratio is biased towards L. helveticus, a situation that tends to maximize hybridization rate (Hubbs, 1955; Randler, 2002; Secondi et al., 2006). Here again, limited introgression outside syntopic sites is expected.
Materials and methods
Sampling strategy
In order to estimate the overall hybridization rate, we collected DNA samples from 37 ponds. One pond, Liré, was sampled two years in a row (2007 and 2008). Five sites, from three and two distinct areas for L. helveticus and L. vulgaris, respectively, were located in allopatry. In addition, 32 ponds from 10 distinct areas were sampled in the sympatric zone. Among these, 10 ponds were allotopic and 22 were syntopic. In total, 928 L. helveticus and 344 L. vulgaris were sampled (Figure 1 and Table 1).
In order to analyse genotypic and phenotypic introgression at a local scale, we increased our sampling effort in one area. Twenty out of the 32 sympatric ponds were located in the Loire river valley and adjacent plateaus between Saumur and Nantes (Figure 1 and Table 1).
Genetic analysis
Microsatellite analysis
We analysed DNA from the 928 L. helveticus and 344 L. vulgaris individuals. Individuals were toe- or tailed-clipped according to the capture permit provided for each sampling area. DNA was extracted following Chelex procedures (Walsh et al., 1991). We used six microsatellite markers to estimate introgression level: Th09, Th14, Th27, ThCa14, Tv3Ca9, Tv3Ca19 (Johanet et al., 2009a). PCR amplifications were performed using fluorescent-labelled forward primers and 10-μl reaction volumes containing 10 ng genomic DNA, 0.4 μM of each primer, 0.1 mM of each deoxynucleotide triphosphate, 2.0 mM MgCl2, 0.2 units Taq DNA polymerase (Promega, Charbonnières-les-Bains, France). Reactions were performed in a MJ Research PT200-Tetrad thermal cycler (GMI, Ramsey, MN, USA): 4 min at 95 °C, 35 cycles consisting of 30 s at 95 °C, 30 s at Tm °C (see Johanet et al., 2009a), 30 s at 72 °C, then 5 min at 72 °C. PCR products were separated on an ABI Prism 3130xl DNA Analyser.
We used MICRO-CHECKER to check microsatellite data for null alleles (Oosterhout et al., 2004). Genetic differentiation between species was calculated using FST estimates (Weir and Cockerham, 1984), and tested by permutation test using GENETIX (Montpellier, France, Belkhir et al., 2004). We ran a factorial correspondence analysis (CA) using GENETIX (Belkhir et al., 2004) on ponds with no a priori species identity. CA is particularly well suited for hybridization studies as it generally extracts between-species differentiation on the first axis, and within-species population differentiation on further axes (Daguin et al., 2001). Interestingly, CA simultaneously expresses the genetic variation in the data set and the respective contributions of each allele to the variation.
Hybrid identification
We used two Bayesian-Markov Chain Monte Carlo (MCMC) methods to detect hybrids, NEWHYBRIDS (Anderson and Thompson, 2002, avalaible at http://ib.berkeley.edu/labs/slatkin/eriq/software/software.htm) and STRUCTURE (Pritchard et al., 2000, avalaible at http://pritch.bsd.uchicago.edu/structure.html). Neither of them requires a priori assignment of alleles to taxonomic entities and both use markers that are not necessarily fixed in either parental species. Moreover, each method can accommodate null alleles.
STRUCTURE (Pritchard et al., 2000) was used to estimate the most likely number of genetic entities (that is, clusters) in the sample and then individual admixture proportions. The number of clusters in our data set was estimated by pooling all samples and calculating the probability that each individual belong to one of K clusters. We assumed that K ranged from one to six clusters, considering the fact that pure parents and various classes of hybrids (F1, F2 and backcrosses) can represent distinct genetic entities. Individual admixture proportions (that is, ancestry estimates) were calculated by assuming an admixture model (that is, allowing the genetic composition of individuals to be a mixture from different populations), using 10 000 iterations for burn-in and 10 000 iterations for parameter estimation. This was repeated 20 times at each K-value. The most likely K-value was estimated using the method proposed by Evanno et al. (2005). We used a threshold of 90% to assign an individual to one parental group or another (Duputié et al., 2007).
Additionally, we used NEWHYBRIDS (Anderson and Thompson, 2002) to estimate the posterior probability that an individual falls into one of the six following categories: pure parental (L. helveticus or L. vulgaris), F1, F2 and backcrosses to one parental species or another. The whole data set was analysed after 10 000 iterations. We assigned an individual to one of the parental groups when its inferred proportion of genes belonging to the group exceeded 90% (Smulders et al., 2008). Otherwise, individuals were considered as hybrids of first or further generation.
We tested for the effect of species and sex on hybrid frequency using log-linear analyses (StatSoft, 2007). This method analyses multifactor cross-tabulations by partitioning the variation in frequency data into the different factors, testing for interactions in a way similar to the analysis of variance.
Mitochondrial analysis of putative hybrids
We genotyped mitochondrial DNA of putative hybrids to identify their maternal parents. We used BioEdit (Hall, 1999) to align sequences from the control region of Lissotriton boscai (Martínez-Solano et al., 2006) and Primer 3 (Rozen and Skaletsky, 2000) to design primers for the amplification of an approximately 300 bp sequence. PCR conditions were the same as for Lissotriton boscai, with 0.4 μM of each primer (DloopboscaiF: CAGCTGGTATGAATCTATGA, DloopboscaiR: CTTAGAAAAACTAGCTCCGG). The annealing temperature was 51 °C. PCR products were then separated on a non-denaturing polyacrylamide gel to screen single-strand conformational polymorphism (Fujita and Silver, 1994). Amplified fragments were mixed with an equal volume of sample buffer, denatured at 95 °C for 5 min, and cooled on ice. A volume of 10 μl of the mix was deposited on a 8% non-denaturing gel containing 14 ml of 37.5% polyacrylamide-1% bisacrylamide mixture, 7 ml of glycerol 0.5 X, 7 ml of TBE 5 X (450 mM Tris-borate, 10 mM EDTA at pH 8) and 42 ml of H2O. Electrophoresis ran at a constant voltage of 8 W for 16 h in l X TBE electrophoresis buffer. Silver staining revelation followed Creste et al. (2001). Gels were washed in 2 l cold (4 °C) fixing solution (10% absolute ethanol, 1.5% acetic acid) for 10 min. Washed gels were soaked for 3 min in 2 l cold solution with 1.5% nitric acid, then washed and impregnated for 10 min in a 2 l cold solution of 1 g l−1AgNO3. Gels were rinsed quickly (20 s) once with 2 l distilled H2O and developed in a 30 g l−1 Na2CO3 solution with 40% formaldehyde until the bands appeared with a sufficient intensity.
Introgression analysis
Even low hybridization, as expected in Lissotriton (Arntzen et al., 1998), can lead to substantial introgression (Goodman et al., 1999). Here we used a method that is appropriate when hybridization is rare. It separates the contribution of ancestral polymorphism from current hybridization to apparent introgression, allowing no strictly diagnostic markers (Barton, 2000). Microsatellite alleles at a single locus (excluding those with null alleles) were clustered in two groups according to their first axis coordinate on the CA. We defined two species-specific synthetic alleles: Lh, characteristic of L. helveticus-like individuals and Lv, characteristic of L. vulgaris-like individuals. They also corresponded to the alleles inferred by STRUCTURE to be characteristic of clusters 1 and 2, respectively, when we fixed K=2 clusters. Synthetic alleles were used to compute between-genome deviations from Hardy–Weinberg equilibrium (κ1,1), and within-genome pairwise associations (κ0,2, linkage disequilibrium) following the method described by Barton (2000), using MATHEMATICA 5.0 (Wolfram Research, Inc, Champaign, IL, USA). We expected between-genome and within-genome pairwise associations to be at their maximum values when two syntopic taxa do not hybridize. As recommended by Barton (2000), the likelihood of different nested models was estimated. We assumed first that the population is in Hardy–Weinberg and linkage equilibria, then allowed for pairwise associations between-genomes (Hardy–Weinberg disequilibrium), within-genomes (linkage disequilibrium) or both, and finally for higher-order associations. The same sequence of models was repeated allowing different contributions of each locus to the association. The most appropriate model was chosen using the Akaike information criterion under which model 2 is more likely than model 1 if log(L2)-2n >log(L1), wherein n is the number of additional parameters used in model 2 relative to model 1. A 95% confidence interval was estimated using a random walk algorithm with parameter T (temperature of the simulated annealing) initially set up to 1 (Barton, 2000). This method does not need to use reference populations that represent parental gene pools.
We calculated for each pond the difference between the observed value for Hardy–Weinberg disequilibrium and linkage disequilibrium compared with their theoretical maximum value by the formula κmaximum−κobserved/κmaximum. In order to detect an influence of sex-ratio on interspecific matings, we then tested for a relation between Hardy–Weinberg and linkage equilibria and sex-ratio in each population using a linear model using R 2.6.1 (R Development Core Team, 2007, http://www.r-project.org/).
Phenotypic analysis
In order to phenotypically detect hybridization, we measured a sample of 560 adults from the Loire river area composed of 389 L. helveticus (181 males and 208 females) and 171 L. vulgaris (59 males and 112 females). Adult newts were caught in ponds from March to May in the year 2006 and 2007 using dipnets (capture and release permit provided by Préfectures du Maine-et-Loire and Loire-Atlantique). All newts were anaesthetized before manipulation by immersion in Tricaine methane sulphonate (0.2 g l−1), MS222, for duration of 5–10 min. Measurements were taken the same day or the day after capture. Four characters associated with body size and secondary sexual trait development were measured to the nearest 0.01 mm using a digital calliper: snout-to-vent length, maximum tail height, cloaca width and filament length (for details on the measurement method, see Secondi et al., 2007). Characters were measured twice and the mean value of the two measurements was recorded. We also scored the throat pattern by coding ‘0’ for a clear throat (typical of L. helveticus) to ‘4’ for a completely marbled throat (typical of L. vulgaris). These traits are known or likely to discriminate the two species (Griffiths and Mylotte, 1988; Johanet et al., 2009b). Individuals were subsequently released to their original capture sites.
Principal component and discriminant analyses were performed using STATISTICA (StatSoft, 2007) to determine whether morphological clusters could be detected without any a priori hypothesis of group membership. Sexes were treated separately because of sexual dimorphism for all traits. To check for a relationship between genotype and phenotype we tested for a correlation between individuals scores in principal component analyses (phenotypic score) and CA (genotypic score) for each sex and species using linear mixed-effects models with population as a random effect in R 2.6.1.
Results
Genetic analysis
Hybrid identification
Polymorphism levels of the six microsatellite markers ranged from 12 to 35 alleles per locus with an average of 25 alleles in both species. Two microsatellite loci, Th27 and Th14, were fully diagnostic. The proportion of private alleles was 41% in L. helveticus and 39% in L. vulgaris. After analysis with MICRO-CHECKER two loci showed null alleles (Th27 and Th09). Genetic differentiation was significant (F̂ST=0.263, P=0.000) between the two taxa. Consistently, factorial CA on microsatellite data supported the genetic distinctiveness of L. helveticus and L. vulgaris by separating two clusters along the first axis (Figure 2). The large eigenvalue (analogous to a partial FST estimate) of the first axis, 25.13%, was indicative of a strong differentiation among samples. Samples from each species formed a scatterplot; however, the two 99% ellipsoids partially overlaid. A few individuals of each species, according to their phenotype, were located on the edge or within the ellipsoid of the other species. Interestingly, variation on both axes was much larger in L. vulgaris (x-values ranging from 0 to 2200 approximately) than in L. helveticus (x-values ranging from ∼−800 to 0).

Scatterplot of the two first axes of a factorial correspondence analysis based on multilocus analysis of 927 Lissotriton helveticus (open circles) and 343 L. vulgaris (black filled triangles) sampled over the breeding ranges of the two species. The dotted and plain ellipsoids represent 99% confidence intervals for L. helveticus and L. vulgaris, respectively. Grey open squares represent L. helveticus-like hybrids and grey filled squares L. vulgaris-like hybrids. The two asterisks indicate the L. helveticus-like hybrid with a L. vulgaris mitochondrial DNA and the L. vulgaris-like hybrid with a L. helveticus mitochondrial DNA.
Cluster analysis using STRUCTURE revealed that the most likely number of populations K in our sample was two (Appendix 1). 98.1% of all individuals (1248/1272) were classified as L. helveticus or L. vulgaris. The remaining individuals (24/1272) were considered as hybrids. Figure 3 illustrates the distribution of individuals according to their ancestry estimates (posterior probability of belonging to L. vulgaris). It shows a clear segregation of allopatric individuals. All individuals with intermediate genotypes come from the sympatric zone, 21 from syntopic sites and three from allotopic sites.

Distribution of ancestry estimates from STRUCTURE analysis (proportion of individual genomes derived from Lissotriton vulgaris) in L. helveticus and in L. vulgaris in different ecological contexts.
According to our classification criterion and in agreement with STRUCTURE assignments, NEWHYBRIDS assigned 98.3% of all individuals (1251/1272) to a parental group, L. helveticus or L. vulgaris, and 1.7% (21/1272) of all individuals to the remaining four groups. NEWHYBRID assigned none of these individuals to any one of the hybrid classes with a high probability. For instance, no F1 was detected and five individuals were assigned with a probability higher than 30% to the F2 class, all of them possessing private alleles from the two species. Results may reflect the inability of NEWHYBRID to identify the correct hybrid class, a known weakness of this method when several generations of backcross occur (Anderson and Thompson, 2002). Focusing on the two diagnostic loci, five hybrid individuals clearly showed two private alleles for one species at one diagnostic locus and one L. helveticus allele plus one L. vulgaris allele for the other diagnostic locus, suggesting that recombination occurred.
Outputs given by the two Bayesian clustering methods were congruent. According to our criterion, a total of 25 hybrids were identified by NEWHYBRIDS and STRUCTURE. Twenty of them were identified by both methods using the same assignment threshold for posterior probabilities. Among the five remaining individuals, four were identified by STRUCTURE only and one by NEWHYBRIDS only. These five individuals were detected by both methods when we raised the assignment threshold to 95%.
We used these data to compute overall hybridization rate and its geographical variation. All hybrids were captured in sympatry from five distinct areas. Hybridization rate did not vary largely between areas: 2.4% (18/743 in 12 ponds) in Pays-de-la-Loire, 3.4% (1/29 in one pond) in Pas-de-Calais, 1.0% in Oise (1/97 in one pond), 2.6% (1/39 in one pond) in Sussex and 3.8% (1/26 in one pond) in Lorraine. All but three hybrids originated from syntopic ponds. Overall hybridization rate was 1.7%. It was 2.9% if we only consider syntopic sites. Note that we excluded from this computation individuals from site Liré 2008 (three hybrids were detected) that is a replicate of Liré 2007 (three hybrids were detected the year before).
Introgression analysis
Models assuming pairwise associations between genomes (Hardy–Weinberg disequilibrium) and within genomes (linkage disequilibrium) were the most appropriate. Pairwise associations of alleles at a locus between genomes, κ1,1 and across loci within a genome, κ0,2 were presented as a function of synthetic Lv allele mean frequency in Figure 4. κ1,1 and κ0,2 could not be estimated for all 13 allotopic L. helveticus sites, the Swedish allopatric L. vulgaris site and six syntopic sites because at least one allele was fixed in these sites. Theoretical values for these parameters were used in Figure 4. No departure from maximum disequilibrium was observed among L. vulgaris from allopatry (Norwegian population). Sixteen sites showed significant deviation both from Hardy–Weinberg, κ1,1 and linkage, κ0,2 disequilibrium maximum values, indicating that interspecific gene exchanges occurred. All of them were syntopic. Each sample of the site of Liré (2007 and 2008) was treated as separate populations and both showed significant introgression. Introgression was thus detected in 73% (16/22) of all syntopic sites.

Pairwise associations within-genomes, κ0,2 (corresponding to an average pairwise linkage disequilibrium) and between-genomes, κ1,1 (corresponding to an average Hardy–Weinberg disequilibrium) estimated following the method of organized as a function of Lissotriton vulgaris allele frequency. Samples of L. helveticus in allotopy are represented by white circles, sample of L. vulgaris in allotopy are represented by black triangle. Samples in syntopy are represented by grey rectangulars for sites in Pays-de-Loire and by grey diamonds for sites elsewhere in Europe. Theoretical maximum values of κ0,2 and κ1,1 are represented by curves. Vertical bars represent 95% confidence intervals.
Sex, species and hybridization
Among the 25 newts identified as hybrids, nine were phenotypically classified as L. helveticus (six males and three females) and 16 as L. vulgaris (5 males and 11 females). Hybrid frequency was 4.8 times lower in L. helveticus-like individuals (1.0%) than in L. vulgaris-like individuals (4.7%). We used a log-linear model to test the effect of species and sex on hybrid frequency. The full model (model with both effects included) gave a significantly better fit to the data than the reduced model (model with no effect) (ΔLog-likelihood=7.8484, χ2=15.6968, df=3, P<0.0013). We observed more hybrids in L. vulgaris-like than in L. helveticus-like individuals (χ2=16.569, df=1, P<0.0001). No effect of sex (χ2=0.325, df=1, P=0.568) and no interaction between species and sex (χ2=0.499, df=1, P=0.478) were detected.
Interestingly, we observed significant contrasting sex-ratio values between our main study area (Loire river valley) and the other sites (Appendix 2). Sex-ratios were more significantly female biased in the Loire valley than elsewhere in Europe for L. helveticus (pMann−Whitney=0.001) and for L. vulgaris (pMann−Whitney=0.004). The difference was particularly strong for L. vulgaris. These differences are independent of the stages of the breeding season as our sampling scheme is composed of several spatio-temporal replicates. We tested the relationship between deviations from maximum values of κ1,1 and κ0,2, and site sex-ratio. Deviation from κ0,2 was correlated with sex-ratio in L. helveticus (F=5.019, P=0.047), whereas deviation from κ1,1 was correlated with sex-ratio in L. vulgaris (F=5.440 P=0.042). According to the coefficient signs (−0.550 and −1.038), the more interspecific gene exchanges occurred in the population (that is, the more linkage or Hardy–Weinberg disequilibrium decreased), the more sex-ratio was biased to the detriment of males.
Mitochondrial DNA analysis
Mitochondrial DNA analysis was carried out on 168 allopatric individuals and the 25 hybrids. Based on the 168 reference samples, we found two species-specific haplotypes for each species. Mitochondrial DNA matched phenotype for all allopatric individuals and for most hybrids. Mismatch between phenotype and mitochondrial DNA was observed only in one out of nine L. helveticus-like hybrids, and one out of 16 L. vulgaris-like hybrids (see Figure 2). These two individuals were males. Mismatch rate did not differ between species (Fisher exact test, P=0.581).
Phenotypic analysis
The Principal component analyses based on morphological traits (Figure 5) clearly separated males and females of the two species as 99% confidence ellipses did not overlap. Note that species discrimination was supported by Axis 1 for males (47.28% of phenotypic variation) and Axis 2 for females (axis 1=48.96%, axis 2=31.27%). Species discrimination was significant for each sex (males: Wilk’s-Lambda=0.0758, P<0.0000; females: Wilk’s-Lambda=0.0240 P<0.0000). Discriminant function analysis classified all males and females to the initial group they have been assigned to by observers.

Scatterplot of the two first axes of a principal component analysis based on phenotypic traits measured in (a) 240 males and (b) 320 females of Lissotriton helveticus (white circles) and L. vulgaris (black triangles) sampled in the Loire valley. Note that dotted and plain ellipsoids represent 99% confidence intervals for L. helveticus and L. vulgaris, respectively.
We tested for the relationship between phenotype and genotype using principal component analyses -scores and CA-scores of individuals on axes responsible for species discrimination. We observed neither significant relationship in male and female L. helveticus, nor in male L. vulgaris. In contrast, we detected a significant positive relationship in female L. vulgaris (coefficient=225.4640, standard error (SE)=72.2812, df=4, F=9.7298, P=0.0024). This result means that the more genetically intermediate L. vulgaris females were, the more they resembled to L. helveticus females.
Discussion
In a situation of large geographical and ecological overlap, we quantified introgression between two newt species, tested for the effect of sex and species and sought to explain the maintenance of taxonomic integrity by studying different areas and syntopic contexts across their distribution.
Hybrid frequency
We used two assignment methods, NEWHYBRIDS and STRUCTURE, that identified hybrids left undetected by morphometry. No F1 hybrids were found but different levels of admixture were observed. Because of the low number of diagnostic loci (Vähä and Primmer, 2006) and the limits of the Bayesian models (only recent hybrids can be found, Anderson and Thompson, 2002), assignment methods are not expected to detect all hybrids. Nevertheless, results given by NEWHYBRIDS and STRUCTURE were congruent and the same individuals were identified as hybrids provided assignment threshold was slightly elevated for NEWHYBRIDS.
Our estimation of hybrid frequency in adults was two to three times higher (1.7%) than the frequency of alien genes (0.07%) observed earlier in larvae (Arntzen et al., 1998). The discrepancy between the two studies may reflect either better survival of hybrids to metamorphosis or properties of the markers used (microsatellites vs allozymes). The first hypothesis is not confirmed by experimental crossings (Spurway and Callan, 1960). The second hypothesis is more plausible. A high variance among markers in levels of differentiation between species is expected when the presence of semipermeable genetic barriers to gene exchange is suspected (Bierne et al., 2003). The small sample size of markers used in both studies (two enzymatic diagnostic markers and six microsatellites) may explain this high variance between markers and therefore the apparent discrepancy.
We found no morphological evidence of hybridization between the two newts. Phenotype distribution was strongly bimodal and discriminant analysis classified all individual in the group they have been assigned to in the field. It seems unlikely that morphology alone would be enough to survey the hybridization process as it is often difficult to discriminate introgressed from pure individuals in the wild (Naisbit et al., 2003; Thomas et al., 2008). Results highlighted the fact that most hybrids remain phenotypically undetected. Males’ sexual morphology is highly differentiated whereas females are very similar. It is thus not surprising that only hybrid males have been observed by others in the field (Griffiths et al., 1987; Arntzen et al., 1998; Beebee et al., 1999; Schlüpmann et al., 1999), even if both species are among the most common newts in Europe.
Species and sex asymmetric introgression
Mitochondrial analysis revealed that both maternal lineage L. helveticus and L. vulgaris occurred in hybrids. Consistently, nuclear gene flow was bidirectional between the two species. However, we found a significant asymmetric pattern of nuclear introgression as the proportion of hybrids or introgressed individuals was five times higher in L. vulgaris than in L. helveticus. The range of scores on the first CA axis was also much larger for L. vulgaris than for L. helveticus, which is consistent with the fact that a larger proportion of L. vulgaris was involved in heterospecific crosses than L. helveticus. Several ecological factors (relative abundance, reproductive phenology) and selective factors could explain this pattern but none of them have been rigorously tested here. Still, we observed that relative abundance was often biased as we found more L. helveticus than L. vulgaris in our study area. Unbalanced abundances facilitate the formation of heterospecific pairs and favour asymmetrical heterospecific mating rates (Arnold et al., 1993; Randler, 2002). This factor may have a major effect on the dynamics of the hybrid zone. We also observed a genotype–phenotype relationship in L. vulgaris females but not in L. helveticus females or males of both species. The rationale for this pattern is not straightforward as it could result from an actual asymmetric relationship if phenotypes of both species responded differentially to hybridization, or simply reflects a higher frequency of L. vulgaris-like hybrids. In any case, the current situation suggests that hybrid L. vulgaris-like females would more frequently backcross with L. vulgaris males than with L. helveticus males and therefore contribute to maintain an asymmetrical hybridization pattern.
The negative relationships between disequilibria deviations from their maximum values (κ) and sex-ratio for each species indicates that in sites where hybridization occurred, sex-ratio was significantly female-biased. Even if we randomized temporal sampling across the different sites, we cannot discard the possibility that temporal variation inside the breeding season biased sex-ratio estimates (Chadwick et al., 2006). However, the positive correlation between genotypic and phenotypic scores found in L. vulgaris females and not in males is also suggestive of selection against hybrid males. Haldane's rule could apply here as males are the heterogametic sex in Lissotriton (Hillis and Green, 1990). Sex-specific postmating barriers may be associated with a decrease in viability and in fitness of hybrid males. However this remains to be rigorously tested. Another way to interpret the negative relationship between sex-ratio and introgression level in populations is that the more biased the sex-ratio is, the more mismating may occur. According to Randler (2002), scarcity of conspecifics facilitates hybridization in general, no matter which sex is the rare one. Operational sex-ratio could also affect hybridization; mismating may be higher early in the season as female newts are less selective in early breeding (Gabor and Halliday, 1997).
Extent of introgression
Overall, about 1.7% of adults had probabilities of admixture with each species. Hybrid frequency fluctuated around this value in several areas of the sympatric zone. Sampling effort differed between those areas but is unlikely to explain such low values. Thus, hybridization appears as a phenomenon occurring over a large proportion of species’ breeding ranges. Tests for multilocus Hardy–Weinberg and linkage equilibrium provided additional evidence for the existence of mixing between the two taxa. We found significant deviations from theoretical maximum values in about 70% of syntopic sites. According to our data, introgression was thus common in syntopy but did not leak out into allotopy. The result is striking as allotopic and syntopic sites in the Loire river study area alternate along the valley on a short geographical scale (Johanet et al., 2009b). We may not have had enough power to accurately detect all hybrids as suggested by NEWHYBRIDS and STRUCTURE output. Method for estimating disequilibria is also awkwardly sensitive to allele fixation as only one fixed allele impairs computation. Thus, we may have underestimated actual hybrid frequency and introgression. Nevertheless, both approaches consistently indicate that introgression is present in sites where the two species physically co-occur. We cannot discard the occurrence of introgression in allotopy but its level is certainly lower than in syntopy. Overall, our results are consistent with the maintenance of species integrity, as observed on phenotypic traits, in spite of recurrent interspecific gene flow.
Introgression dynamics in broad hybrid zones
Our study showed that a moderate level of hybridization does not compromise genetic integrity of species with broad geographic overlap that are regularly found in syntopy. The contact zone between the two newts is old (that is, after the last glaciation, Spurway and Callan, 1960) and yet both taxa remain well genetically differentiated in the large fraction of the contact zone that we sampled. The apparent restriction of hybrids to syntopic sites raises questions about the factors that impede the diffusion of alleles to allotopy and more largely about the dynamics of introgression in broad hybrid zones. Owing to the age of these taxa, it is possible that most neutral or advantageous alleles have already spread into the other species distribution and that markers currently showing some degree of diagnosticity are subject to selection, directly or via strong linkage to another gene.
However, one has to consider introgression in the general context of intraspecific gene flow for each species. In L. helveticus, the number of detected hybrids is small relative to the total population size and allotopic sites are more abundant than syntopic ones. Thus, genetic drift may wipe out introgressed alleles from allotopic sites. A recent study by Petit and Excoffier (2009) proposes that introgression is impaired when intraspecific gene flow is high. Accordingly, net gene flow from allopatric or allotopic areas reduces the frequency of alien alleles that are then more easily lost by chance. Hybrid frequency is not particularly small in the L. helveticus–L. vulgaris system. Still, hybridization rate does not seem high enough for introgression to spread. Interestingly, L. helveticus is quite ubiquitous and relatively abundant in our study site. It tends to occupy a wider niche and more breeding sites than L. vulgaris. In such a context, the frequency of hybrid effective migrants needs to be high enough to counter the homogenizing effect of intraspecific gene flow.
The maintenance of species integrity between broadly sympatric species is usually the result of strong post- and pre-zygotic barriers so that the few hybrids produced are considered as evolutionary dead-ends. However, other processes may prevent introgression without the elevation of particularly strong reproductive barriers. The relative abundance of species and the proportion of syntopic sites over the total number of sites (allotopic and syntopic) are two major factors that may strongly condition the demographic component of broad hybrid zones, and set the upper limit to introgression dynamics. The spatial arrangement of allotopic and syntopic sites will determine net relative intra- and inter-specific gene flow and for this reason should also be considered. The strength of the effects generated by these different factors may create demographic barriers that impair the diffusion of alien genes outside syntopic sites. Their efficiency is probably reinforced for aquatic species that breed in discrete habitats such as ponds or lakes. Modelling would likely shed light on the process of genome interaction between taxa hybridizing in broad hybrid zones. The emphasis should be put on the effects and interactions of drift, relative abundance and relative occurrence on introgression dynamics.
References
Anderson EC, Thompson EA (2002). A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160: 1217–1229.
Arnold ML, Hamrick JL, Bennett BD (1993). Interspecific pollen competition and reproductive isolation in Iris. J Hered 84: 13–16.
Arntzen J, Wijer PD, Jehle R, Smit E, Smit J (1998). Rare hybridization and introgression in smooth and palmate newts (Salamandridae: Triturus vulgaris and T. helveticus). J Zool Syst Evol Research 36: 111–122.
Arntzen JW (1986). Note sur la coexistence d′espèces sympatriques de tritons du genre Triturus. Bull Soc Herp Fr 37: 1–8.
Baack EJ, Rieseberg LH (2007). A genomic view of introgression and hybrid speciation. Curr Opin Genet Dev 17: 513–518.
Babik W, Szymura JM, Rafinski J (2003). Nuclear markers, mitochondrial DNA and male secondary sexual traits variation in a newt hybrid zone (Triturus vulgaris × T. montandoni). Mol Ecol 12: 1913–1930.
Barton NH (2000). Estimating multilocus linkage disequilibria. Heredity 84: 373–389.
Barton NH, Hewitt GM (1985). Analysis of hybrid zones. Ann rev ecol syst 16: 113–148.
Beebee TJC, Rowe G, Arntzen JW (1999). Identification of newts (Triturus sp.) and their hybrids using molecular methods. J Zool Lond 249: 43–47.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004). GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations, Laboratoire Génome, Populations, Interactions; CNRS UMR 5000; Université Montpellier II, Montpellier (France).
Benazzi M (1957). Sulla ibridazione fra Triturus helveticus e Tr. vulgaris. Boll Zool 24: 235–242.
Bierne N, Daguin C, Bonhomme F, David P, Borsa P (2003). Direct selection on allozymes is not required to explain heterogeneity among marker loci across a Mytilus hybrid zone. Mol Ecol 12: 2505–2510.
Borge T, Lindroos K, Nádvorník P, Syvänen AC, Sætre GP (2005). Amount of introgression in flycatcher hybrid zones reflects regional differences in pre and post-zygotic barriers to gene exchange. J Evol Biol 18: 1416–1424.
Chadwick EA, Slater FM, Ormerod SJ (2006). Inter- and intraspecific differences in climatically mediated phenological change in coexisting Triturus species. Glob Chang Biol 12: 1069–1078.
Cogălniceanu D (1994). Experimental hybridation within the Triturus vulgaris species-group (Amphibia, Caudata). Rev Roum Biol 39: 145–150.
Cooley JR, Marshall DC, Hill KBR, Simon C (2006). Reconstructing asymmetrical reproductive character displacement in a periodical cicada contact zone. J Evol Biol 19: 855–868.
Coyne JA, Orr HA (2004). Speciation. Sinauer Associates: Sunderland.
Creste S, Tulmann Neto A, Figueira A (2001). Detection of single sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol Biol Report 19: 299–306.
Daguin C, Bonhomme F, Borsa P (2001). The zone of sympatry and hybridization of Mytilus edulis and M. galloprovincialis, as described by intron length polymorphism at locus mac-1. Heredity 86: 342–354.
Denoël M, Ficetola GF (2007). Landscape-level thresholds and newt conservation. Ecol Appl 17: 302–309.
Denoël M, Ficetola GF (2008). Conservation of newt guilds in an agricultural landscape of Belgium: the importance of aquatic and terrestrial habitats. Aquat Conserv Mar Freshw Ecosyst 18: 714–728.
Duputié A, David P, Debain C, McKey D (2007). Natural hybridization between a clonally propagated crop, cassava (Manihot esculenta Crantz) and a wild relative in French Guiana. Mol Ecol 16: 3025–3038.
Duvernell DD, Schaefer JF, Hancks DC, Fonoti JA, Ravanelli AM (2007). Hybridization and reproductive isolation among syntopic populations of the topminnows Fundulus notatus and F. olivaceus. J Evol Biol 20: 152–164.
Endler JA (1977). Geographic variation, speciation, and clines. Princeton University Press: Princeton.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 14: 2611–2620.
Fujita K, Silver J (1994). Single-strand conformational polymorphism. Genome Res 4: 137–140.
Fuller RC (2008). Genetic incompatibilities in killifish and the role of environment. Evolution 62: 3056–3068.
Gabor CR, Halliday TR (1997). Sequential mate choice by multiply mating smooth newts: females become more choosy. Behav Ecol 8: 162–166.
Goodman SJ, Barton NH, Swanson G, Abernethy K, Pemberton JM (1999). Introgression through rare hybridization: a genetic study of a hybrid zone between red and sika deer (genus Cervus) in Argyll, Scotland. Genetics 152: 355–371.
Griffiths RA (1986). Feeding niche overlap and food selection in smooth and palmate newts, Triturus vulgaris and T. helveticus, at a pond in mid-Wales. J Anim Ecol 55: 201–214.
Griffiths RA (1987). Microhabitat and seasonal niche dynamics of smooth and palmate newts, Triturus vulgaris and T. helveticus, at a pond in mid-Wales. J Anim Ecol 56: 441–451.
Griffiths RA, Mylotte VJ (1988). Observations on the development of the secondary sexual characters of male newts, Triturus vulgaris and T. helveticus. J Herp 22: 476–480.
Griffiths RA, Roberts JM, Sims S (1987). A natural hybrid newt, Triturus helveticus x T. vulgaris, from a pond in mid-Wales. J Zool Lond 213: 133–140.
Haldane JBS (1922). Sex ratio and unisexual sterility in hybrid animals. J Genet 12: 101–109.
Hall TA (1999). BIOEDIT: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98.
Harrison RG (1986). Pattern and process in a narrow hybrid zone. Heredity 56: 337–349.
Harrison RG (1993). Hybrid zones and the evolutionary process. Oxford University Press: New York.
Hey J (2006). Recent advances in assessing gene flow between diverging populations and species. Curr Opin Genet Dev 16: 592–596.
Hillis DM, Green DM (1990). Evolutionary changes of heterogametic sex in the phylogenetic history of amphibians. J Evol Biol 3: 49–64.
Höbel G, Gerhardt HC (2003). Reproductive character displacement in the acoustic communication system of green tree frogs (Hyla cinerea). Evolution 57: 894–904.
Howard DJ (1986). A zone of overlap and hybridization between two ground cricket species. Evolution 40: 34–43.
Hubbs CL (1955). Hybrdization between fish species in nature. Syst Zool 4: 1–20.
IUCN (2008). http://www.iucnredlist.org.
Jang Y, Won YJ, Choe JC (2009). Convergent and divergent patterns of morphological differentiation provide more evidence for reproductive character displacement in a wood cricket Gryllus fultoni (Orthoptera: Gryllidae). BMC Evol Biol 9: 27.
Johanet A, Picard D, Garner T, Dawson D, Morales-Hojas R, Jehle R et al. (2009a). Characterization of microsatellite loci in two closely related Lissotriton newt species. Cons genet 10: 1903–1906.
Johanet A, Secondi J, Pays O, Pagano A, Lodé T, Lemaire C (2009b). A case of reproductive character displacement in female palmate newts (Lissotriton helveticus). C R Biol 332: 548–557.
Martin L, Cruzan M (1999). Patterns of hybridization in the Piriqueta caroliana complex in central Florida: evidence for an expanding hybrid zone. Evolution 53: 1037–1049.
Martínez-Solano I, Teixeira J, Buckley D, García-París M (2006). Mitochondrial DNA phylogeography of Lissotriton boscai (Caudata, Salamandridae): evidence for old, multiple refugia in an Iberian endemic. Mol Ecol 15: 3375–3388.
Naisbit RE, Jiggins CD, Mallet J (2003). Mimicry: developmental genes that contribute to speciation. Evol Develop 5: 269–280.
Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004). Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Petit RJ, Excoffier L (2009). Gene flow and species delimitation. Trends Ecol Evol 24: 386–393.
Pritchard J, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Randler C (2002). Avian hybridization, mixed pairing and female choice. Anim Behav 63: 103–119.
Roberts JM (1990). Reproductive isolation in Smooth and Palmate newts (Triturus vulgaris and T. helveticus). PhD Thesis, Open University, Milton Keynes.
Roques S, Sévigny JM, Bernatchez L (2001). Evidence for broadscale introgressive hybridization between two redfish (genus sebastes) in the North-west Atlantic: a rare marine example. Mol Ecol 10: 149–165.
Rozen S, Skaletsky H (2000). Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds)Bioinformatics Methods and Protocols in the Series Methods in Molecular Biology. Humana Press: Totowa, pp 365–386.
Rubidge EM, Taylor EB (2004). Hybrid zone structure and the potential role of selection in hybridizing populations of native westslope cutthroat trout (Oncorhynchus clarki lewisi) and introduced rainbow trout (O. mykiss). Mol Ecol 13: 3735–3749.
Schlüpmann M, Weber G, Lipscher E, Veith M (1999). Nachweis eines freinlandbastardes von Teichmolch (Triturus vulgaris) und Fadenmolch (Triturus helveticus). Zeit Feldherp 6: 203–217.
Secondi J, Aumjaud A, Pays O, Boyer S, Montembault D, Violleau D (2007). Water turbidity affects the development of sexual morphology in the palmate newt. Ethology 113: 711–720.
Secondi J, Faivre B, Bensch S (2006). Spreading introgression in the wake of a moving contact zone. Mol Ecol 15: 2463–2475.
Secondi J, Johanet A, Pays O, Cazimajou F, Djalout Z, Lemaire C (2010). Olfactory and visual species recognition in newts and their role in hybridization. Behaviour (in press).
Sites JWJ, Marshall JC (2003). Delimiting species: A Renaissance issue in systematic biology. Trends Ecol Evol 18: 462–470.
Smulders MJM, Beringen R, Volosyanchuk R, Vanden Broeck A, Van Der Schoot J, Arens P et al. (2008). Natural hybridisation between Populus nigra L. and P. x canadensis Moench. Hybrid offspring competes for niches along the Rhine river in the Netherlands. Tree Genet Genomes 4: 663–675.
Spurway H, Callan HG (1960). The vigour and male sterility of hybrids between the species Triturus vulgaris and T. helveticus. J Genet 57: 84–118.
StatSoft (2007). STATISTICA (data analysis software system), version 8.0.
Steinfartz S, Vicario S, Arntzen JW, Caccone A (2007). A Bayesian approach on molecules and behavior: Reconsidering phylogenetic and evolutionary patterns of the Salamandridae with emphasis on Triturus newts. J Exp Zool B Mol Dev Evol 308: 139–162.
Thomas DT, Ahedor AR, Williams CF, DePamphilis C, Crawford DJ, Xiang QY (2008). Genetic analysis of a broad hybrid zone in Aesculus (Sapindaceae): Is there evidence of long-distance pollen dispersal? Int J Plant Sci 169: 647–657.
Turner TL, Hahn MW, Nuzhdin SV (2005). Genomic islands of speciation in Anopheles gambiae. PLoS Biol 3: 1572–1578.
Vähä JP, Primmer CR (2006). Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol 15: 63–72.
Walsh PS, Metzger DA, Higuchi R (1991). Chelex® 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513.
Wambreuse P, Bels V (1984). Analyse qualitative et quantitative de la parade sexuelle du triton palmé Triturus helveticus (Razoumowsky 1798). Cah Ethol Appl 4: 193–218.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Weisrock DW, Macey JR, Ugurtas IH, Larson A, Papenfuss TJ (2001). Molecular phylogenetics and historical biogeography among salamandrids of the ‘true’ salamander clade: Rapid branching of numerous highly divergent lineages in Mertensiella luschani associated with the rise of Anatolia. Mol Phylogenet Evol 18: 434–448.
Acknowledgements
We thank all the researchers and naturalists who provided us with DNA samples (cf Table 1 and also C Miaud, P Joly, C Jackob, D Henrio, FJ Diego-Rasilla, M Lužnik and D Schmeller). We are grateful to J Dobigeon, T Duret, R Robin and E Beslot for their help in the field and during morphometric measurements and to S Sourice and V Hardouineau for technical assistance. We also thank N Bierne for his help in analysis of cumulants, S Caillault for computation of breeding range areas, J Endler, JW Arntzen, CA Hipsley and three anonymous reviewers for helpful comments and critical improvements on the paper. We thank Région Bretagne for their financial support for analyses in the Plateforme OUEST Genopole (INRA, Rennes) and Solène Couedel who welcomed us. This study was funded by Région Pays-de-la-Loire, DIREN Pays-de-la-Loire and Angers Loire Métropole.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Appendices
Appendix 1
Graphical representation of ΔK is calculated as ΔK=m∣L′(K)∣/s(L(K)) for each K value (Evanno et al., 2005). The modal value of this distribution is two clusters.

Appendix 2
Boxplots of sex-ratio in Lissotriton helveticus (Lh) and L. vulgaris (Lv) from different sites (n) in Pays-de-Loire and elsewhere in Europe. Sites not considered in genetic analyses were included.

Rights and permissions
About this article
Cite this article
Johanet, A., Secondi, J. & Lemaire, C. Widespread introgression does not leak into allotopy in a broad sympatric zone. Heredity 106, 962–972 (2011). https://doi.org/10.1038/hdy.2010.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.144
Keywords
This article is cited by
-
Inconsistent estimates of hybridization frequency in newts revealed by SNPs and microsatellites
Conservation Genetics (2024)
-
Mate preference, species recognition and multimodal communication in heterogeneous environments
Evolutionary Ecology (2015)
-
Extensive mitochondrial introgression in North American Great Black-backed Gulls (Larus marinus) from the American Herring Gull (Larus smithsonianus) with little nuclear DNA impact
Heredity (2014)
-
An ultraviolet signal generates a conflict between sexual selection and species recognition in a newt
Behavioral Ecology and Sociobiology (2014)
-
Habitat-Dependent Species Recognition in Hybridizing Newts
Evolutionary Biology (2014)
