
Abstract
Lentiviral vectors (LVs) provide unique opportunities for the development of immunotherapeutic strategies, as they transduce a variety of cells in situ, including antigen-presenting cells (APCs). Engineering LVs to specifically transduce APCs is required to promote their translation towards the clinic. We report on the Nanobody (Nb) display technology to target LVs to dendritic cells (DCs) and macrophages. This innovative approach exploits the budding mechanism of LVs to incorporate an APC-specific Nb and a binding-defective, fusion-competent form of VSV.G in the viral envelope. In addition to production of high titer LVs, we demonstrated selective, Nb-dependent transduction of mouse DCs and macrophages both in vitro and in situ. Moreover, this strategy was translated to a human model in which selective transduction of in vitro generated or lymph node (LN)-derived DCs and macrophages, was demonstrated. In conclusion, the Nb display technology is an attractive approach to generate LVs targeted to specific cell types.
Similar content being viewed by others


Background
Dendritic cells (DCs) and macrophages are imperative for the activation of antigen-specific T cells.1, 2 Consequently, these antigen-presenting cells (APCs) have been studied as targets in immunotherapeutic strategies for the treatment of cancer and infectious diseases. Numerous strategies for antigen delivery to APCs have been developed.3 Of these, lentiviral vectors (LVs) are particularly appealing, as LVs can accommodate large gene inserts, provide long-term expression and deliver foreign genes to dividing as well as non-dividing cells.4 The latter characteristic makes them exceptionally interesting for transduction of terminally differentiated cells, such as DCs.4 Several publications have reported on the in vitro transduction of DCs with high efficiency and little to no toxicity. It was moreover demonstrated that these DCs retain their maturation potential and induce therapeutic immune responses.5 In addition, LVs have been evaluated as an off-the-shelf vaccine, demonstrating that LVs deliver their cargo to DCs, simultaneously activating DCs through pathogen recognition receptors, such as protein kinase R6, 7 and Toll-like receptors,8, 9, 10, 11 upon which the transduced DCs migrate to lymphoid organs where they stimulate strong antigen-specific immune responses.12, 13, 14
Despite their extensive pre-clinical use, translation of LVs to the clinic is still in its early days.15 Engineering LVs targeted to APCs will advance the translation of LVs from bench to bedside. Several groups are actively working on strategies to facilitate LV transduction to specific immune cells by replacing the commonly used VSV envelope glycoprotein by a cell-specific alternative. An example is the use of the measles virus H- and F-proteins to direct LVs to B and T lymphocytes.16, 17, 18, 19 With regard to APC-specific transductional targeting, the use of MHC II-specific single-chain antibodies (scFv) has been extensively studied. Some examples are: (1) N-terminal insertion of a MHC II-specific scFv peptide into VSV.G,20 (2) fusion of an MHC II-specific scFv to an amphotropic murine leukemia virus glycoprotein21 and (3) a chimeric measles virus H-protein, which is mutated for binding to hemagglutinin, but incorporates a MHC II-specific scFv.22 However, the use of chimeric glycoproteins often has a negative effect on the LV stability and/or transduction efficiency. An alternative strategy to target APCs was proposed by the group of Yang et al.23 who targeted the DC-specific molecule DC-SIGN by the use of an engineered Sindbis virus glycoprotein.23 Despite these efforts, it remains difficult to develop LVs that allow transduction of specific APC subsets, without hampering their stability. Recently, an elegant approach was proposed to improve the specificity of retroviral vectors.24 Herein, the natural budding mechanism of retroviruses is exploited to incorporate specific molecules in the viral surface. Consequently, these determine the retroviral tropism. Chandrashekran et al.24 demonstrated that retroviral vectors, produced in an ecotropic producer cell line that over-expresses stem cell factor on its cell membrane, were able to preferentially transduce c-kit-expressing human stem cells. Yang et al.25 further demonstrated that this approach can also be applied for LVs, in this case using an anti-CD20 antibody to mediate specific transduction of B cells. As molecular cloning of classic antibodies or fragments offers serious challenges, alternatives have been explored. One of them is the use of antibodies generated by members of the Camelidae (that is, dromedaries, camels and llamas), which produce a unique class of antibodies composed of two identical heavy chains as opposed to the conventional (four-chain) antibody repertoire.26 The antigen-binding part of the molecule is composed of only one single variable region, termed camelid heavy chain antibody VH or Nanobody (Nb). These Nbs offer many advantages.27 First, although Nbs can be in vivo matured through immunization and share the high-binding affinity and specificity of antibodies, their single-domain nature allows easy cloning and selection of antigen-specific Nbs and drastically reduces the required size of the library that needs to be constructed and screened. Second, the recombinant nature of Nbs allows interesting possibilities at the level of molecular biological manipulations, such as sequence modification, transfer of the antigen specificity and affinity from one Nb to another.28 Finally, as Nbs can be genetically fused to other proteins, it should be possible to present them on the cell membrane of a producer cell line; thus, generating LVs that incorporate a cell-specific Nb in their envelope during budding as described above. We previously raised several Nbs against mouse bone marrow-derived DCs.29 Of these, Nb DC2.1 was shown to target in vitro generated immature and mature DCs, as well as macrophages.29 Therefore, this Nb was used in the present study to develop the Nb display technology and deliver a proof-of-principle on the use of Nbs to target LVs to specific cell types of mouse and human origin.
Results
The Nb display technology allows production of high titer LVs
In this study, we developed a strategy based on the advantageous characteristics of LVs and Nbs to transductionally target LVs to specific cell types. This innovative strategy is called the Nb display technology. Herein recognition of the target cell and subsequent fusion of the target cell membrane with the viral membrane are mediated by two separate proteins, the Nb and VSV.GS,30 respectively. As we are interested in exploiting LVs for immunotherapeutic purposes and since we previously identified Nb DC2.1 as a Nb that specifically binds APCs, in particular DCs and macrophages, we decided to use this Nb to establish a proof-of-concept.29 As a negative control we used Nb BCII10, which binds to subunit 10 of the β-lactamase BcII enzyme of Bacillus cereus.31
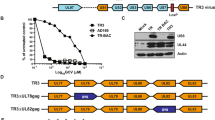
First, we evaluated whether this approach allows the production of LVs with high stability, hence, high titer, as this is a minimal pre-requisite to use LVs for in situ transduction of target cells, such as APCs. To that end, the production of LVs displaying Nb and VSV.GS was compared with the production of VSV.G pseudotyped LVs, as the latter are considered the standard for comparison.32 To do this producer cells stably expressing Nb DC2.1 or BCII10 on their cell membrane were generated. Therefore, human embryonic kidney cells (HEK) 293T cells were transduced with LVs encoding membrane bound Nb DC2.1 or BCII10, respectively. These cells were used to generate VSV.GS pseudotyped LVs. Non-modified HEK 293T cells were used to generate VSV.G pseudotyped LVs. We compared the transfection of Nb expressing versus non-modified HEK 293T cells by flow cytometry (Figure 1a and Supplementary Figure 2, n=6), demonstrating high expression of the transgene (Thy1.1 or tNGFR) on all transfected HEK 293T cells, as well as high expression of the Nbs (myc tag) on Nb-modified HEK 293T cells. Next, we measured the reverse transcriptase (RT) content of the LVs (Figure 1b), demonstrating high levels of RT in the LV preparations. Comparison of the RT content with the titer as determined in flow cytometry of VSV.G pseudotyped LVs revealed that 1 ng RT correlated with 2.5 × 104 TU (transducing units), demonstrating that the titers obtained for Nb displaying LVs are suitable for in vivo applications (n=6). To verify the presence of the Nbs on the LV particles, we first performed an ELISA in which an anti-VHH antibody was used to capture the Nb displaying LVs, after which binding of these LVs was demonstrated using an anti-Thy1.1 detection antibody (Figure 1c, n=3). As a final quality control, we performed western blot on known amounts of LVs (5 ng RT) to assess the amount of VSV.GS and Nbs in the viral preparations. To that end, we used an anti-hemagglutinin A (HA) antibody, which binds to the HA tag present within both the VSV.GS and Nb (Figure 1d, n=4). We moreover determined the density of the western blot signals in order to determine the ratio of Nbs versus VSV.GS, demonstrating that this ratio was not only stable intra-, but also inter LV preparations (Figure 1e, n=4).

Production of Nb displaying LVs. (a) Non-modified HEK 293T cells or HEK 293T cells stably expressing Nb BCII10 or DC2.1 were used to produce LVs pseudotyped with VSV.G or VSV.GS, respectively. Three days after transfection of these cells with the VSV.G or VSV.GS, gag/pol and transgene encoding plasmids, we evaluated the expression of the transgene (Thy1.1 or tNGFR), as well as the Nb (myc tag) by flow cytometry. Non-transfected HEK 293T cells served as a control. The flow cytometry dot plots demonstrate high expression of the transgene (y axis) in all transfected cells (VSV.G, BCII10 and DC2.1) and high expression of Nbs (x-axis) on the Nb-modified HEK 293T cells (BCII10 and DC2.1). One representative experiment is shown (n=6). (b) To compare the LV preparations we determined their RT content. The graph depicts the amount of RT (ng RT/μl) in the LV preparations. Each dot represents one LV stock, the horizontal line shows the mean (n=6). (c) An ELISA involving anti-VHH and anti-Thy1.1 as capture and detection antibodies, respectively, was used to demonstrate the incorporation of Nbs into the surface of Thy1.1 encoding LVs. A serial dilution of LVs was applied (5, 2.5 and 1.25 ng RT). The graph depicts the OD-values detected. One representative experiment is shown (n=3). (d) Western blot was performed as a quality control of the LVs. After separation on a 15% sodium dodecyl sulphate-polyacrylamide gel and transfer to a nitrocellulose membrane, the Nbs (±25 kDa) and VSV.GS (±15 kDa), which both contain an HA epitope tag, were detected with an anti-HA antibody. One representative experiment is shown (n=4). (e) The density of the western blot signals was determined using the Photocapt MW software and used to determine the ratio of Nbs/VSV.GS on the LVs. This ratio is shown in the graph, in which each dot represents one LV stock and the horizontal line shows the mean (n=4).
LVs displaying Nb DC2.1 target mouse APCs
On the basis of the knowledge that Nb DC2.1 binds specifically to mouse DCs and macrophages, we hypothesized that unlike VSV.G pseudotyped LVs, Nb DC2.1 displaying LVs should exclusively transduce DCs and macrophages.29 To evaluate this hypothesis, we first transduced mouse fibroblasts (NIH 3T3), T (EL4 cells) and B (A20 cells) cells, macrophages (RAW246.7 cells) and bone marrow-derived DCs in vitro with VSV.G pseudotyped or Nb BCII10 or DC2.1-displaying LVs encoding Thy1.1 at multiplicity of infection 10. Flow cytometry was performed 72 h after transduction and demonstrated Thy1.1 expression in all cell types upon transduction with VSV.G pseudotyped LVs. In contrast, none of the cell types evaluated were transduced when incubated with BCII10 displaying LVs. More importantly, upon transduction with Nb DC2.1 displaying LVs, we observed Thy1.1 expression by DCs and macrophages, but not by fibroblasts, B or T cells (Figure 2a and Supplementary Figure 3, n=3). We furthermore demonstrated that the observed transduction was Nb-mediated, as pre-incubation of in vitro generated DCs with an excess of Nb DC 2.1, but not Nb BCII10, abrogated their transduction by Nb DC2.1 displaying LVs (Figure 2b, n=2).

Selective transduction of mouse DCs and macrophages by Nb DC2.1 displaying LVs. (a) Mouse NIH 3T3 fibroblasts, A20 B lymphoma and EL4 T lymphoma cells, RAW 264.7 macrophages and bone marrow-derived DCs were mock transduced or transduced with VSV.G pseudotyped or Nb BCII10 or DC2.1 displaying LVs (multiplicity of infection 10). Flow cytometry was performed 72 h later to evaluate transgene, Thy1.1, expression. The histograms demonstrate Thy1.1 positivity in the evaluated cell types. The tinted, blue, black and red histogram, represent mock transduced cells or cells transduced with Nb BCII10 displaying LVs, VSV.G pseudotyped LVs or Nb DC2.1 displaying LVs, respectively. One representative experiment is shown (n=3). (b) Mouse bone marrow-derived DCs were pre-incubated with an excess of Nb BCII10 or DC 2.1, after which these were transduced with Thy1.1 encoding LVs displaying the respective Nbs. Expression of Thy1.1 was determined 72 h later. The dot plots demonstrate the Thy1.1 expression by DCs upon mock transduction (control), transduction with VSV.G pseudotyped LVs, LVs displaying Nb BCII10 or DC2.1 when pre-incubated with Nb BCII10 or DC2.1. One representative experiment is shown (n=2). (c) To evaluate the targeting strategy in vivo, we first administered 105 TU of FLuc encoding LVs to the inguinal LN of C57BL/6 mice. In vivo bioluminescence imaging was performed 36 h later to obtain bioluminescent pseudo-color images, in which high luminescence (a measure for the amount of FLuc positive cells) is shown in red and weak luminescence in blue. The color scale underneath the images represents the LUT or Look up Table, and correlates the luminescence to an absolute amount of counts (light units). The pseudo-color images were superimposed on gray-scale photographs of the mice (n=3). (d) To confirm transduction, hence, proviral integration in the genome of LN cells, we performed a nested PCR on the genomic DNA isolated from these LNs. Subsequently, the PCR fragments were separated using a 1.2% agarose gel. Lanes 1–7 depict the 1 kb DNA ladder, water control of the first (W1) and second (W2) PCR, and the PCR amplification products of genomic DNA isolated from LNs injected with VSVG pseudotyped LVs or LVs pseudotyped with VSV.GS and displaying Nb BCII10 or DC2.1 (n=3). (e, f) LVs encoding Thy1.1 were administered (106 TU) to the inguinal LN in order to track LV transduced cells. Analyses were performed 36 h later on single cell suspensions prepared from these LNs. In order to evaluate Thy1.1 expression in macrophages, myeloid DCs and lymphoid DCs (together conventional DCs), pDCs, B and T cells the LN cells were stained with the antibody directed against Thy1.1 in combination with antibodies directed against CD11b F4/80, CD11c CD8α, CD11c CD8α, CD11c B220, CD19 and CD3, respectively. The dot plots in (e) depict the cell populations of which the Thy1.1 expression is shown in the graphs in (f). One representative experiment is shown (n=3).
As our main goal is to use targeted LVs for in situ modification of cells, we next evaluated the specificity of the Nb DC2.1 displaying LVs in vivo. As Nb DC2.1 targets LVs to APCs in vitro, we decided to deliver the targeted LVs to the inguinal lymph node (LN) of C57BL/6 mice, as LNs have a relative high concentration of both DCs and macrophages.33, 34, 35 First, we administered LVs (105 TU) encoding FLuc. Thirty-six hours later, in situ transduction was evaluated using in vivo bioluminescence imaging (Figure 2c), demonstrating luminescence when VSV.G pseudotyped or Nb DC2.1 displaying LVs were administered, but not when Nb BCII10 displaying LVs were administered (n=3). We next performed a nested PCR on genomic DNA isolated from these LNs, confirming that absence of luminescence upon delivery of Nb BCII10 displaying LVs was truly because of the lack of transduction and that the luminescence observed with VSV.G pseudotyped and Nb DC2.1 displaying LVs was owing to genuine transduction (Figure 2d, n=3). These data demonstrate that Nb DC2.1 displaying LVs transduce cells in situ, however, they do not indicate specificity. Therefore, C57BL/6 mice with a Thy1.2 background were injected in the inguinal LN with 106 TU of LVs encoding Thy1.1. Thirty-six hours later, LNs were isolated, reduced to a single cell suspension and characterized. Cells were stained with an anti-Thy1.1 antibody in combination with antibodies against CD11b and F4/80, CD11c and CD8α, CD11c and B220, CD3 or CD19 to evaluate Thy1.1 expression by macrophages, conventional DCs (cDCs), plasmacytoid DCs (pDCs), T or B cells, respectively (Figure 2e, n=3). Flow cytometry demonstrated a similar transduction pattern of the evaluated LVs as observed in vitro, that is, VSV.G pseudotyped LVs transduced all cell types evaluated, whereas Nb BCII10 displaying LVs transduced none. More importantly, Nb DC2.1 displaying LVs were transductionally targeted to macrophages, cDCs and pDCs. Moreover, the transduction of cDCs, which are thought to mediate immune responses upon LV transduction, was enhanced when targeted LVs were used when compared with VSV.G pseudotyped LVs (Figure 2f and Supplementary Figure 4, n=3).
LVs displaying Nb DC2.1 target human APCs
The experiments described above demonstrate efficient gene delivery to APCs in vivo. This greatly enhances the therapeutic potential of LV-based immunotherapeutic strategies as targeting reduces the risk of insertional mutagenesis and off-target effects. However, to translate this strategy to the clinic, targeting of human APCs has to be evaluated. As it was demonstrated that Nb DC2.1 also binds human APCs (data not shown), we next translated the mouse data described above to a human model. First, we transduced in vitro cultured human fibroblasts, monocyte-derived macrophages and DCs, as well as blood-derived B and T cells with Thy1.1 encoding LVs at an multiplicity of infection of 10. Flow cytometry performed 72 h later demonstrated that the DC2.1 displaying LVs specifically targeted human DCs and macrophages, whereas Nb BCII10 displaying LVs did not; VSV.G pseudotyped LVs again transduced all cell types evaluated (Figure 3a and Supplementary Figure 5, n=3). Next, we generated single cell suspensions of human LNs and transduced these cells with Thy1.1 encoding LVs. Thirty-six hours later, the cells were stained for Thy1.1, as well as CD14 and CD11b, CD11c and BDCA-3, CD123 and BDCA-2, CD3 or CD19, to evaluate Thy1.1 expression by macrophages, myeloid DCs, pDCs, T cells or B cells, respectively (Figure 3b, n=3). These experiments demonstrate that VSV.G pseudotyped LVs transduced all cell types evaluated, whereas Nb BCII10 displaying LVs did not. Importantly, selective transduction of macrophages, myeloid DCs and pDCs was observed upon transduction with DC2.1 displaying LVs (Figure 3c and Supplementary Figure 6, n=3). Comparable to what was observed in mice, Nb DC2.1 displaying LVs appeared to transduce myeloid DCs more efficiently when compared with macrophages and pDCs, whereas VSV.G pseudotyped LVs transduced these cell types at equal efficiency. These data demonstrate that the Nb display technology can be applied to target human APCs.

Selective transduction of human DCs and macrophages by DC2.1 displaying LVs. (a) Human fibroblasts, blood-derived B and T cells, in vitro generated macrophages and bone marrow-derived DCs were mock transduced or transduced with VSV.G pseudotyped, or Nb BCII10 or DC2.1 displaying LVs (multiplicity of infection 10). Flow cytometry was performed 72 h later to evaluate transgene (Thy1.1) expression. The histograms demonstrate Thy1.1 positivity in the evaluated cell types. The tinted, blue, black and red histogram, represent mock transduced cells or cells transduced with Nb BCII10 displaying LVs, VSV.G pseudotyped LVs and Nb DC2.1 displaying LVs, respectively. One representative experiment is shown (n=3). (b, c) Single cell suspensions prepared from human LNs were transduced in vitro with Thy1.1 encoding LVs (multiplicity of infection 10). In order to evaluate Thy1.1 expression in macrophages, myeloid DCs, pDCss, B and T cells, these cells were co-stained with the anti-Thy1.1 antibody and antibodies directed against CD11b CD14, CD11c BDCA-3, CD123 BDCA-2, CD19 and CD3, respectively. The flow cytometry graphs in panel C depict Thy1.1 expression; the corresponding cell populations are shown in the histograms displayed in panel (b). The tinted, blue, black and red histogram represent, mock transduced cells or cells transduced with Nb BCII10 displaying LVs, VSV.G pseudotyped LVs or Nb DC2.1 displaying LVs, respectively. One representative experiment is shown (n=3).
Discussion
Several vaccination strategies with great potential have been developed to treat diseases, such as cancers, chronic infections and autoimmune disorders. One of these is the use of LVs to deliver cancer, viral or autoimmune antigens together with immune-modulating molecules to APCs, which subsequently induce the appropriate immune response, that is, immunity or tolerance, respectively.36 Optimization of the safety and efficiency of the LVs used to modify APCs could make them an even more powerful tool for developing novel treatment modalities against various diseases.
In this report, we present the Nb display technology to generate LVs targeted to APCs. This approach exploits the natural budding mechanism of LVs to incorporate a binding-defective, but fusion-competent envelope glycoprotein derived from VSV.G30 together with a membrane bound APC-specific Nb. To our knowledge, there have been no prior reports on the use of Nbs for targeting of LVs. Furthermore, we are the first to demonstrate (1) production of high titer APC-targeted LVs, (2) confirmation of in vivo transduction of LN cells upon delivery of targeted LVs by in vivo bioluminescence imaging and (3) specific transduction of human LN DCs and macrophages.
Production of LVs at a high titer is a pre-requisite for their application in vivo. As, we wanted to compare the titers of broad tropism LVs to those of Nb displaying LVs, we first generated Nb-expressing producer cells. As a consequence, the LV production was based on the classical three-plasmid transfection for both LV types. We observed no cytotoxicity because of the expression of Nbs on the producer cells, which underscores the suitability of Nbs as small non-toxic molecules for incorporation on LVs. Furthermore, we demonstrate that the Nb-expressing cell lines allow the production of LVs at similar titers as VSV.G pseudotyped LVs, the LV type against which other LVs are compared.32 The ability to generate LVs at high titers is an important advantage of our strategy. Although the group of Funke et al.37 was able to improve both titer and selectivity by pseudotyping LVs with wild-type measles virus glycoproteins, most alternative strategies don’t report on this phenomenon. In general, titers and specificity of pseudotypes, including N-terminal insertion of MHC II-specific scFv20 to the VSV.G, murine leukemia virus-A21 or mutated measles virus H-protein,22 or the use of a modified Sindbis virus envelope glycoprotein,23 were lower compared with those of the Nb displaying LVs described in this study.
To address the transduction specificity, we evaluated the transduction profile of Nb DC2.1 displaying LVs on mouse as well as human APCs. We confirmed Nb-dependent and APC-specific transduction of Nb DC2.1 displaying LVs on murine cell lines and in vitro generated DCs. We showed in vivo transduction with the Nb DC2.1 displaying LVs after intranodal injection using in vivo bioluminescence imaging and confirmed these results by nested PCR. Although in vivo bioluminescence imaging has been previously applied to prove specific transduction of tumor cells,38 we are the first to report on successful bioluminescent images of in vivo transduced APCs with targeted LVs. Previous attempts to evidence targeted APC transduction in vivo were made by subcutaneous injection of FLuc encoding LVs. The lack of luminescence in these experiments was ascribed to the sparse distribution of skin-derived DCs, which was beyond the sensitivity of the applied imaging method.23 Phenotypic characterization of the in situ transduced LN cells demonstrated that the entry of Nb DC2.1 displaying LVs was limited to macrophages, cDCs and pDCs. Importantly, the transduction of myeloid DCs, which are thought to mediate immune responses upon LV transduction,1 was enhanced when these targeted LVs were used.
Selective transduction of DC2.1 displaying LVs was moreover evidenced on human in vitro generated APCs, as we demonstrated transduction of macrophages and DCs, but not fibroblasts, B or T cells. More importantly, we were able to confirm these data on human LN-derived cells. This may facilitate translation of the data obtained in mice to a relevant human model. Interestingly, similar to the mouse data, Nb DC2.1-displaying LVs were more efficient in transducing human myeloid DCs than human macrophages or pDCs. The difference in transduction efficacy of the Nb displaying LVs can be interpreted in several ways. First, binding of the Nb-displaying LVs and their subsequent fusion with the target cell membrane are mediated by two different molecules, a binding Nb and a fusogenic VSV.GS molecule, which is opposed to only one protein (VSV.G) in broad tropism LVs. However, this also holds true for LVs pseudotyped with the measles virus envelope glycoprotein for which several groups reported on high transduction efficacy.18, 19, 22, 37 Alternatively, the reduced transduction efficacy can be explained by the binding process of the LVs on the target cell. Most cells heavily express the receptor to which VSV.G binds,39 whereas the expression of the antigen, recognized by Nb DC2.1, is limited to DCs and macrophages. Moreover, the identity of this antigen is unknown. We can’t exclude variation in the expression of the antigen depending on the DC and macrophage subtype and its activation status. The successful work of several groups targeting CD20 or MCH II is suggestive for this hypothesis as these molecules are highly expressed on the respective target cells.20, 21, 22, 40 The latter explanation is not detrimental to the proposed work, but indicates that it is critical to identify the best target antigen and binding Nb with regard to targeting.
It will be critically important to extend the current pre-clinical studies applying targetable LVs into an off-the-shelf immunotherapy approach for clinical applications. We believe that the described approach provides an important step toward this goal as it tackles major concerns such as off-target transduction and the risk on insertional mutagenesis.41 We believe that the latter is severely reduced with our approach as APCs are cells with a relatively short life span, and as transformation is a multistep process,42 oncogenesis is unlikely to occur.
Finally, one of the major advantages of this system is that it can be easily applied to target different cell types. As mentioned in the introduction the generation, selection and molecular cloning of Nbs is straightforward. Moreover, Nbs can be generated against any cell type without the need for prior knowledge of a cell-specific marker, which can be a challenge in itself. Consequently, the Nb display technology further enhances the potential of LVs as a widely used gene delivery vehicle for fundamental research, functional genomics and gene therapy purposes.
Materials and methods
Mice, human LNs and cell cultures
Six to 12-week-old C57BL/6 female mice (Thy1.2) were purchased from Harlan (Horst, The Netherlands). Animals were handled according to the institutional guidelines and experiments were approved by the Ethical Committee for use of laboratory animals of the VUB (Protocol no 10-214-1, date 31-03-2010). Approval to use LNs from organ donors was obtained from the institutional review board (Protocol no BUN14320108848, date 26-06-2010).
HEK 293T, NIH 3T3 cells (mouse fibroblasts), RAW264.7 cells (mouse leukemic macrophage cell line), EL4 cells (mouse T-lymphoma cell line) and A20 cells (mouse B-lymphoma cell line) were cultured as recommended by the American Type Culture Collection (ATCC, Rockville, MD, USA). The generation of mouse bone marrow-derived and human monocyte-derived DCs, as well as the isolation of B and T cells from peripheral blood was performed as previously described.5 Monocytes were selected by adherence and cultured in the presence of M-CSF in order to generate human macrophages.
Single cell suspensions were prepared from murine and human LNs. For the latter, the procedure described for preparation of single cell suspensions from mouse LNs was adapted.8 Briefly, human LNs were injected with phosphate buffered saline (Lonza, Verviers, Belgium) containing Collagenase III (100 U ml−1, Sigma-Aldrich, Bornem, Belgium) and DNase I (32.5 U ml−1, Sigma-Aldrich). The LNs were immersed in 500 μl of phosphate buffered saline containing 0.5% human AB serum (PAA Laboratories, Linz, Austria) and incubated at 37 °C, 5% CO2 for 30 min, after which they were tamped with a plunger of a 3-cc syringe. A single cell suspension was obtained by passing the cells through a 70-μm cell strainer on a 15-ml tube. The cells were cultured at 1 × 106 cells ml−1 in X-VIVO 15 medium (BioWhittaker, Walkersville, MD, USA) containing 1% human AB serum.
LV production
Plasmids
The packaging plasmid pCMVΔR8.9 and VSV.G encoding plasmid pMD.G were a gift from Dr D Trono (University of Geneva). The plasmid pUB6-VSV.GS, which encodes the binding-defective, but fusion-competent VSV.G was described by Zhang et al.30 The VSV.GS is schematically represented in Supplementary Figure 1A. The plasmids encoding Thy1.1 (pSIN-Thy1.1) or Firefly Luciferase (FLuc, pHR trip CMV luc2-Ires-tNGFR SIN) were previously described.8 The sequence encoding Nb BCII10 or DC2.129 was cloned into the phage display vector, pHEN6c as a NcoI-Eco91I (BstEII) fragment. Subsequently the cloning sites SacII and SalI were introduced in the Nb sequence by PCR, after which the Nbs were cloned as a SacII-SalI fragment in the vector pDISPLAY (Invitrogen, Paisley, UK), resulting in pDISPLAY-Nb BCII10 and pDISPLAY-Nb DC2.1. As a consequence, the Nb-encoding sequence is fused at the N-terminus to the mouse Igκ chain leader sequence and at the C-terminus to the platelet derived growth factor receptor transmembrane domain, to direct the Nb to the secretory pathway and subsequently anchor it to the plasma membrane. The Nbs expressed from this vector further contain the HA and myc epitopes (used for western blot and flow cytometry, respectively). A PCR to introduce the SpeI and EcoRI cloning sites into the membrane bound version of Nb BCII10 and DC2.1 was performed, after which this fragment was cloned into the backbone pHR’ vector, using these restriction sites.5
Virus production
In order to prepare LVs, HEK 293T cells were plated at 15 × 106 cells per 175 cm2. These were transfected the following day using polyethyleneimine (Polysciences, Eppelheim, Germany) with 15, 30 and 45 μg of the envelope, gag/pol and transgene encoding plasmids, respectively. LV-containing supernatant was collected the following 3 days and concentrated by ultracentrifugation (1000 × g) as previously described.5
Virus characterization
The colorimetric RT assay (Roche, Vilvoorde, Germany) was used to determine the amount of RT in the concentrated LV preparation.43 Comparison of the RT content with the titer, as determined in flow cytometry, of VSV.G pseudotyped LVs revealed that 1 ng RT correlated with 2.5 × 104 TU. ELISA was performed following standard procedures using an anti-VHH and an anti-Thy1.1 antibody (Becton Dickinson, BD, Regenbogen, Belgium) as capture and detection antibody, respectively. Western blot was performed on LVs (5 ng RT). Viral proteins were separated on a 15% sodium dodecyl sulfate-polyacrylamide gel, transferred to a nitrocellulose membrane, after which Nbs and VSV.GS, which contain an HA tag, were detected with an anti-HA antibody (Sigma-Aldrich) and a horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Santa Cruz Biotechnology, Heidelberg, Germany) as primary and secondary antibody, respectively. The signal density was measured using the Photocapt MW software (Vilber Lourmat, Marne-La-Vallee, France) to determine the ratio between Nbs and VSV.GS on LVs.
Transduction of cells
In vitro transduction of mouse and human fibroblasts, DCs, macrophages, B and T cells, as well as LN cells, was performed at a multiplicity of infection of 10 as previously described.5 To achieve transduction of cells in situ the inguinal LN of C57BL/6 mice was injected with 105–106 TU of LVs resuspended in 10 μl phosphate buffered saline containing 10 μg ml−1 protamine sulphate (LeoPharma, Lier, Belgium). Analyses of in vitro transduced cells was performed 36 (primary cells) or 72 h (cell lines and in vitro generated DCs) after transduction, whereas analyses of in vivo transduced cells was performed 36 h after LV injection.
In vivo bioluminescence imaging
In vivo bioluminescence imaging was performed as previously described to visualize in situ transduction of LN cells by FLuc encoding LVs.8, 44
Nested PCR
Genomic DNA was isolated from LNs injected with FLuc encoding LVs using the QIAamp DNA mini kit (Qiagen, Antwerpen, Belgium). Integrated pro-viruses were detected by nested PCR. The initial PCR was performed on 500 ng of genomic DNA with the forward primer (CPPT 8951) 5′-AGGGGAAAGAATAGTAGACAT-3′ and reverse primer (B2) 5′-ATATGTAA GTACACTGTAGC-3′, using a hot start polymerase mix (Kapa Biosystems, Eke, Belgium) and the following PCR program 95 °C 5′, 35 × (95 °C 15″, 60 °C 15″, 72 °C 30″), 72 °C 7′ and hold at 4 °C. The second PCR was performed on 1 μl of the first PCR reaction with the forward primer (CMV 9551) 5′-CAAATGGGCGGTAGGCGTGTA-3′ and reverse primer (Lenti Rev) 5′-CCTTGTAA GTCATT GGTCCTTAA-3′, using the same enzyme and PCR program.
In vivo tracking of LVs
To evaluate transduction of LN cells by Thy1.1 encoding LVs, mice were killed, the injected LN isolated, after which a single cell suspension was prepared using Liberase TL (Roche). Cells were characterized in flow cytometry.
Flow cytometry
Staining of surface markers was performed as previously described.5 A biotinylated anti-myc antibody (Millipore, Brussels, Belgium) was used to assess the expression of Nbs on Nb modified HEK 293T cells. Phycoerythrin conjugated anti-Thy1.1 (Biolegend, ImTec Diagnostics, Antwerpen, Belgium) or anti-NGFR (BD) antibodies were used to evaluate transgene expression in modified cells. Mouse cells were characterized using: allophycocyanin conjugated antibodies against CD11c (BD) and CD19 (Biolegend), fluorescein isothiocyanate conjugated antibodies against CD11b (BD), peridinin–chlorophyll proteins-Cy5.5 (PerCP-Cy5.5) conjugated antibody against CD8αBD and biotinylated antibodies against F4/80, CD3 and B220 (made in house). Human cells were further characterized using: APC conjugated antibodies against CD11c (BD), CD19 (BD) and CD123 (BD); fluorescein isothiocyanate conjugated antibodies against CD14 (BD) and CD3 (BD); PerCP-Cy5.5 conjugated antibodies against CD11b (BD) and biotinylated antibodies against BDCA-3 and BDCA-2 (Miltenyi Biotec, Leiden, The Netherlands). Biotinylated antibodies were detected with a streptavidin-PerCP-Cy5.5 (BD). Data were collected using a FACSCanto flow cytometer (BD) and analyzed using FACSDiva (BD) or FlowJo software (Tree Star, Inc., Ashland, OR, USA).
References
Breckpot K, Escors D . Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr Metab Immune Disord Drug Targets 2009; 9: 328–343.
Pollard JW . Trophic macrophages in development and disease. Nat Rev Immunol 2009; 9: 259–270.
Breckpot K, Heirman C, Neyns B, Thielemans K . Exploiting dendritic cells for cancer immunotherapy: genetic modification of dendritic cells. J Gene Med 2004; 6: 1175–1188.
Breckpot K, Emeagi PU, Thielemans K . Lentiviral vectors for anti-tumor immunotherapy. Curr Gene Ther 2008; 8: 438–448.
Breckpot K, Dullaers M, Bonehill A, van Meirvenne S, Heirman C, de Greef C et al. Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med 2003; 5: 654–667.
Breckpot K, Emeagi P, Dullaers M, Michiels A, Heirman C, Thielemans K . Activation of immature monocyte-derived dendritic cells after transduction with high doses of lentiviral vectors. Hum Gene Ther 2007; 18: 536–546.
Tan PH, Beutelspacher SC, Xue SA, Wang YH, Mitchell P, McAlister JC et al. Modulation of human dendritic-cell function following transduction with viral vectors: implications for gene therapy. Blood 2005; 105: 3824–3832.
Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S et al. HIV-1 lentiviral vector immunogenicity is mediated by TLR3 and TLR7. J Virol 2010; 84: 5627–5636.
Brown BD, Sitia G, Annoni A, Hauben E, Sergi Sergi L, Zingale A et al. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2007; 109: 2797–2805.
Pichlmair A, Diebold SS, Gschmeissner S, Takeuchi Y, Ikeda Y, Collins MK et al. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J Virol 2007; 81: 539–547.
Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest 2005; 115: 3265–3275.
Dullaers M, Van Meirvenne S, Heirman C, Straetman L, Bonehill A, Aerts JL et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther 2006; 13: 630–640.
Esslinger C, Chapatte L, Finke D, Miconnet I, Guillaume P, Levy F et al. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest 2003; 111: 1673–1681.
He Y, Zhang J, Donahue C, Falo Jr LD . Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 2006; 24: 643–656.
Escors D, Breckpot K . Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Exp (Warsz) 2010; 58: 107–119.
Frecha C, Levy C, Costa C, Negre D, Amirache F, Buckland R et al. Measles virus glycoprotein-pseudotyped lentiviral vector-mediated gene transfer into quiescent lymphocytes requires binding to both SLAM and CD46 entry receptors. J Virol 2011; 85: 5975–5985.
Frecha C, Levy C, Cosset FL, Verhoeyen E . Advances in the field of lentivector-based transduction of T and B lymphocytes for gene therapy. Mol Ther 2010; 18: 1748–1757.
Frecha C, Costa C, Levy C, Negre D, Russell SJ, Maisner A et al. Efficient and stable transduction of resting B lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors. Blood 2009; 114: 3173–3180.
Frecha C, Costa C, Negre D, Gauthier E, Russell SJ, Cosset FL et al. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 2008; 112: 4843–4852.
Dreja H, Piechaczyk M . The effects of N-terminal insertion into VSV-G of an scFv peptide. Virol J 2006; 3: 69.
Gennari F, Lopes L, Verhoeyen E, Marasco W, Collins MK . Single-chain antibodies that target lentiviral vectors to MHC class II on antigen-presenting cells. Hum Gene Ther 2009; 20: 554–562.
Ageichik A, Buchholz CJ, Collins MK . Lentiviral vectors targeted to II MHC are effective in immunization. Hum Gene Ther 2011; 22: 1249–1254.
Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol 2008; 26: 326–334.
Chandrashekran A, Gordon MY, Casimir C . Targeted retroviral transduction of c-kit+ hematopoietic cells using novel ligand display technology. Blood 2004; 104: 2697–2703.
Yang L, Bailey L, Baltimore D, Wang P . Targeting lentiviral vectors to specific cell types in vivo. Proc Natl Acad Sci USA 2006; 103: 11479–11484.
Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB et al. Naturally occurring antibodies devoid of light chains. Nature 1993; 363: 446–448.
Revets H, De Baetselier P, Muyldermans S . Nanobodies as novel agents for cancer therapy. Expert Opin Biol Ther 2005; 5: 111–124.
Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K . General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem 2009; 284: 3273–3284.
De Groeve K, Deschacht N, De Koninck C, Caveliers V, Lahoutte T, Devoogdt N et al. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med 2010; 51: 782–789.
Zhang XY, Kutner RH, Bialkowska A, Marino MP, Klimstra WB, Reiser J . Cell-specific targeting of lentiviral vectors mediated by fusion proteins derived from Sindbis virus, vesicular stomatitis virus, or avian sarcoma/leukosis virus. Retrovirology 2010; 7: 3.
Conrath KE, Lauwereys M, Galleni M, Matagne A, Frere JM, Kinne J et al. Beta-lactamase inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother 2001; 45: 2807–2812.
Cronin J, Zhang XY, Reiser J . Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther 2005; 5: 387–398.
Banchereau J, Steinman RM . Dendritic cells and the control of immunity. Nature 1998; 392: 245–252.
Katakai T, Hara T, Lee JH, Gonda H, Sugai M, Shimizu A . A novel reticular stromal structure in lymph node cortex: an immuno-platform for interactions among dendritic cells, T cells and B cells. Int Immunol 2004; 16: 1133–1142.
Nakamura K, Yamaji T, Crocker PR, Suzuki A, Hashimoto Y . Lymph node macrophages, but not spleen macrophages, express high levels of unmasked sialoadhesin: implication for the adhesive properties of macrophages in vivo. Glycobiology 2002; 12: 209–216.
Goyvaerts C, Kochan G, Escors D, Breckpot K . Dendritic cells and lentiviral vectors: mapping the way to successful immunotherapy. Viral Gene Therapy; pp 309–352. ISBN 978-953-307-539-6.
Funke S, Schneider IC, Glaser S, Muhlebach MD, Moritz T, Cattaneo R et al. Pseudotyping lentiviral vectors with the wild-type measles virus glycoproteins improves titer and selectivity. Gene Ther 2009; 16: 700–705.
Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B et al. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat Med 2005; 11: 346–352.
Coil DA, Miller AD . Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J Virol 2004; 78: 10920–10926.
Ziegler L, Yang L, Joo K, Yang H, Baltimore D, Wang P . Targeting lentiviral vectors to antigen-specific immunoglobulins. Hum Gene Ther 2008; 19: 861–872.
Breckpot K, Aerts JL, Thielemans K . Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther 2007; 14: 847–862.
Fehse B, Roeder I . Insertional mutagenesis and clonal dominance: biological and statistical considerations. Gene Therapy 2008; 15: 143–153.
Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S et al. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol 2010; 84: 5627–5636.
Keyaerts M, Verschueren J, Bos TJ, Tchouate-Gainkam LO, Peleman C, Breckpot K et al. Dynamic bioluminescence imaging for quantitative tumour burden assessment using IV or IP administration of D: -luciferin: effect on intensity, time kinetics and repeatability of photon emission. Eur J Nucl Med Mol Imaging 2008; 35: 999–1007.
Jeetendra E, Robison CS, Albritton LM, Whitt MA . The membrane-proximal domain of vesicular stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J Virol 2002; 76: 12300–12311.
Acknowledgements
We thank Elsy Vaeremans, Petra Roman, Xavier Debaere and Dr Aude Bonehill (VUB) for their help in purifying plasmid DNA and the generation of single cell suspensions from human LNs, respectively. We furthermore thank Profs Axel Bossuyt and Tony Lahoutte for the use of the imaging facilities. This research was performed with the financial support of the Research foundation Flanders (FWO-V), the Agency of Innovation by Science and Technology (IWT-SBO), the Interuniversity Attraction Poles Program (IUAP), the Belgian State-Belgian Science Policy and the research committee of the VUB (OZR). CG, LR and KB are funded by the research committee of the VUB (OZR), the IWT-SBO and the FWO-V, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Gene Therapy website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Goyvaerts, C., De Groeve, K., Dingemans, J. et al. Development of the Nanobody display technology to target lentiviral vectors to antigen-presenting cells. Gene Ther 19, 1133–1140 (2012). https://doi.org/10.1038/gt.2011.206
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2011.206
Keywords
This article is cited by
-
Construction of a T7 phage display nanobody library for bio-panning and identification of chicken dendritic cell-specific binding nanobodies
Scientific Reports (2022)
-
Nanobody: a promising toolkit for molecular imaging and disease therapy
EJNMMI Research (2021)
-
A comprehensive comparison between camelid nanobodies and single chain variable fragments
Biomarker Research (2021)
-
Development of an adenovirus vector vaccine platform for targeting dendritic cells
Cancer Gene Therapy (2018)
-
Antigen-presenting cell-targeted lentiviral vectors do not support the development of productive T-cell effector responses: implications for in vivo targeted vaccine delivery
Gene Therapy (2017)

{kind=link}