
Abstract
Purpose: To prospectively validate a quantitative fluorescent polymerase chain reaction (PCR) assay as a method of rapid prenatal aneuploidy detection for chromosomes 13, 18, 21, X, and Y.
Methods: A commercial quantitative fluorescent PCR kit was validated on 200 known, blinded, prenatal DNA specimens. The kit was then validated prospectively on 1069 amniotic fluid specimens, and the results were compared with the karyotype results and the results of interphase fluorescence in situ hybridization testing, when performed in the course of standard care. Turnaround time was monitored in a subset of the prospective specimens.
Results: The analytical sensitivity and specificity of testing in the validation specimens were 98.9% and 100%, respectively. There were no false positives and a single false negative, a mosaic sex chromosome aneuploidy interpreted as normal. In the prospective study, the analytical sensitivity and specificity were 98% and 100%, respectively. No false positives and a single false negative, again a sex chromosome mosaic, were detected. Overall, 72.5% of all chromosomal anomalies and 87.7% of clinically significant chromosome anomalies were detected by quantitative fluorescent PCR. The average and median turnaround times were 30.5 and 25.1 hours, respectively.
Conclusions: Quantitative fluorescent PCR is a robust and accurate method of rapid prenatal aneuploidy detection.
Similar content being viewed by others

Main
In Canada, invasive prenatal diagnosis is recommended for a number of indications, including abnormal maternal serum screening results, advanced maternal age (≥35 years at the time of delivery), ultrasound anomalies, or a family history of a genetic or chromosomal disorder.1,2
Aneuploidies of chromosomes 13, 18, 21, X, and Y are the most common chromosomal anomalies detected in prenatal specimens, representing approximately 70% of all chromosomal anomalies detected.3,4 Currently, the gold standard for prenatal diagnosis of chromosomal anomalies is karyotype analysis using specimens obtained during either amniocentesis or chorionic villus sample (CVS).5 Because karyotype analysis of prenatal specimens requires a culturing period, karyotype results are typically not available until 10–14 days after the procedure. Furthermore, karyotyping is a labor-intensive and expensive process that does not lend itself easily to automation. Shortage of resources in the cytogenetics laboratory can often contribute to delayed turnaround times for these prenatal specimens. This delay can be very stressful for the patient, especially in high-risk situations or when the pregnancy is already quite advanced. Therefore, a rapid test to rule out the most common chromosomal anomalies in all women undergoing amniocentesis or CVS is desirable.
Rapid prenatal aneuploidy detection by fluorescence in situ hybridization (FISH) has been available in most cytogenetics laboratories for at least 15 years.6 FISH uses fluorescently labeled DNA probes complementary to chromosomes 13, 18, 21, X, and Y, thereby allowing scoring of common aneuploidies in interphase nuclei and serves as a powerful adjunct to classical cytogenetics. This technique has the advantage of not requiring cell culture and results can usually be generated within 24–48 hours of the procedure. Interphase FISH has a sensitivity and specificity for detecting aneuploidies of chromosomes 13, 18, 21, X, and Y that approach 100%,7 and there is a commercially available FISH probe set for in vitro diagnostic use (AneuVysion™; Abbott Molecular, Abbott Park, IL). However, the cost of FISH is often prohibitive. Furthermore, FISH is relatively labor intensive, although less so than karyotyping. Consequently, the high cost of reagents and labor intensity of interphase FISH are prohibitive for widespread use, limiting this option to women at the greatest risk. Most centers in Ontario set minimum risk criteria for offering prenatal FISH, although the exact risk figure used varies from center to center.
Other options for rapid prenatal detection of aneuploidy include quantitative fluorescent polymerase chain reaction (QF-PCR), multiplex ligation-dependent probe amplification, and genomic microarray testing.8,9 Multiplex ligation-dependent probe amplification and genomic microarray testing have been reviewed elsewhere and will not be further discussed in this study.8,10 QF-PCR was developed in Europe, and its use has increased in recent years in a number of different countries in Europe and Asia, although its use in North America is still relatively rare.11–34 QF-PCR involves the relative quantitation of polymorphic markers on each of the chromosomes of interest using fluorescently labeled primers to amplify the markers followed by analysis using capillary electrophoresis. The number of alleles and their relative intensity at each marker can then be used to predict chromosome copy number.9 Several multiplexed assays for prenatal diagnosis have been developed,13,15,16 and commercial kits are also now available. Several large studies have now been published that indicate that QF-PCR is a sensitive and specific method for detecting aneuploidy in prenatal specimens.21,24,30,34 These studies suggest that QF-PCR shows a very high level of concordance with conventional cytogenetic studies (99.6% in the largest study to date) with no false positives and very few false negatives.16,28,30 Considering just abnormal results, QF-PCR detected 92.3% of all anomalies and 95% of clinically significant anomalies.30 It is also highly robust with an amplification failure rate of 0.05–0.09% with markers being informative for 99.95% of specimens.16,30 Interpretation of QF-PCR results was impossible due to extensive maternal cell contamination (MCC) in just 1–1.7% of specimens.28,30
The cost of QF-PCR is a fraction of the cost of FISH or conventional karyotyping. Several recent studies have suggested that QF-PCR could replace karyotyping in low-risk pregnancies21,34–38 and has the potential to reduce the need for karyotyping by as much as 90% when combined with other routinely used tests such as ultrasound.35
In this study, a commercially available QF-PCR kit (Aneufast™; Molgentix SL, Spain) was used for the rapid prenatal diagnosis of aneuploidies for chromosomes 13, 18, 21, X, and Y. The assay was validated on retrospective and prospective amniotic fluid specimens. The QF-PCR results were compared with karyotyping and FISH results to evaluate whether QF-PCR was a viable replacement for FISH as a rapid method of prenatal aneuploidy detection. Three primary research questions were asked in this study: (1) What is the analytical validity (sensitivity, specificity, positive predictive value [PPV], and negative predictive value [NPV]) of QF-PCR testing using the Aneufast™ kit in a series of known, blinded specimens? (2) What is the analytical validity of QF-PCR testing using the Aneufast™ kit in a prospective set of unknown specimens? and (3) Is the turnaround time of QF-PCR testing using the Aneufast™ kit acceptable for a rapid prenatal aneuploidy test?
MATERIALS AND METHODS
Study specimens
Retrospective validation specimens
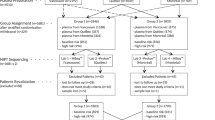
DNA from 200 anonymous amniotic fluid specimens with known karyotype (both normal and abnormal), previously tested in the molecular genetics laboratory at General Lab in Barcelona, Spain, were analyzed by North York General Hospital (NYGH) to validate the QF-PCR assay used in this study. All NYGH staff, including technologists and laboratory directors, were blinded to the results of the specimens until all the analyses were complete.
Prospective patient specimens
Two hospitals with the largest prenatal programs in Ontario participated in this study: NYGH and Mount Sinai Hospital (MSH), both located in Toronto. Between November 2006 and July 2007, all women undergoing amniocentesis at NYGH and MSH were invited to participate in the QF-PCR study. The study was approved by the research ethics boards of both NYGH and MSH. Each patient was given written information about the test and individually counseled by a geneticist/genetic counselor or obstetrician about the risks and benefits of participating in the study. Patients who agreed to participate in the study were asked to sign a consent form. A total of 1069 women were enrolled in the study.
Approximately 1 mL of amniotic fluid required for QF-PCR testing was taken from the specimen obtained for routine cytogenetic analysis. All specimens were coded before submission to facilitate blind testing. All QF-PCR testing was performed in the Molecular Genetics Laboratory at NYGH, which is accredited by Ontario Laboratory Accreditation and has all the appropriate quality assurance measures in place for diagnostic PCR testing. Patients enrolled in the study received their karyotype and FISH (when applicable) results after routine protocols. The karyotype and FISH analyses were performed in the Cytogenetics Laboratory at the hospital in which the specimen was drawn (NYGH or MSH). QF-PCR results were not revealed to the patient.
Specimen preparation
For the retrospective study, DNA from specimens with known karyotypes was prepared at General Lab in Barcelona, Spain, using the same procedure detailed later. The specimens were stored at −20°C for up to 5 years before the study.
For prospective patient specimens, DNA was isolated using Chelex (InstaGene Matrix™, Bio-Rad, Mississauga, Canada) in a rapid isolation technique. The whole extraction procedure was performed in the same tube to reduce the risk of mishandling. Briefly, 1 mL of uncultured amniotic fluid was pelleted by centrifugation for 5 minutes at 13,000 rpm; 50–300 μL of Chelex was added to the pellet depending on the size of the pellet and incubated for 8 minutes at 70°C. The specimen was mixed by vortexing for 10 seconds and incubated for 4 minutes at 95°C. The specimen was mixed by vortexing again for 10 seconds and sedimented by centrifugation for 2 minutes at 13,000 rpm. PCR-ready single-stranded DNA was contained in the supernatant.
For heavily blood-stained amniotic fluid specimens, red cell lysis and washing steps were performed before DNA extraction. Briefly, 1 mL of sterile distilled, deionized water was added to the cell pellet and mixed by vortexing. After incubation at room temperature for 2 minutes, the specimen was pelleted by centrifugation for 5 minutes at 13,000 rpm. Two subsequent wash steps were performed by aspirating the supernatant and adding 1 mL of sterile distilled, deionized water. The pellet was resuspended by vortexing followed by sedimentation by centrifugation for 5 minutes at 13,000 rpm. DNA isolation from the cell pellet was then carried out as outlined earlier.
Quantitative fluorescent polymerase chain reaction
Aneufast™
The Aneufast™ QF-PCR (Molgentix SL, Spain) kit was used in this study. Aneufast™ is a commercially available multiplex QF-PCR kit for rapid prenatal diagnosis of aneuploidies of chromosomes 13, 18, 21, X, and Y. The kit comprises six multiplex marker sets of short tandem repeats (STRs) that can be used for amplification of selected microsatellites and the amelogenin and SRY genes. These multiplex marker sets include two sets (S1 and S2) that include markers from all five chromosomes of interest plus reflex marker sets from each of chromosomes 13 (M13), 18 (M18), and 21 (M21) and a combined X and Y chromosome reflex marker set (MXY). The S1 and S2 marker sets are performed for all specimens, whereas the chromosome-specific reflex marker sets are used to clarify ambiguous results or confirm a positive result. More information on the specific marker composition of the S1 and S2 sets as well as the reflex sets is available from the manufacturer.39
PCR, specimen preparation, and capillary electrophoresis were carried out according to manufacturer's instructions.39
Interpretation
Results were interpreted, according to manufacturer's instructions,39 by comparing the relative amount of fluorescence of multiple alleles of a marker. Normal individuals who are heterozygous will have approximately equal representation of each allele and will, therefore, have a ratio of peak areas of 1:1. An individual who is homozygous at a given marker will be uninformative for that marker. At least two informative markers were required for each chromosome to confirm a result.
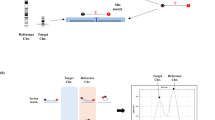
Trisomic specimens have two possible patterns: three different alleles (trisomic triallelic), in which three copies of a chromosome are indicated by the presence of three peaks for corresponding chromosome-specific STRs, all of which have the same fluorescence intensity and a ratio between the areas of 1:1:1 and trisomic diallelic, which produces two unbalanced fluorescent peaks with an area ratio of 2:1 or 1:2 (Fig. 1). Occasionally, homozygosity will be observed rendering the markers uninformative. The diagnosis of trisomy is made if at least two markers on the same chromosome have trisomic patterns.

Representative trisomic STR patterns. Trisomic specimens have two possible patterns: trisomic triallelic, in which three copies of a chromosome are indicated by the presence of three peaks for corresponding chromosome-specific STRs, all of which have the same fluorescence intensity and a ratio between the areas of 1:1:1 (D21S1412) and trisomic diallelic, which produces two unbalanced fluorescent peaks with an area ratio of 2:1 or 1:2 (D21S1809).
Trisomic specimens produce trisomic triallelic and diallelic patterns for all markers on the same chromosome. Triploid specimens produce trisomic diallelic or triallelic patterns for informative STRs on all chromosomes. Tetraploidy cannot be detected by this assay.
X chromosome monosomy (Turner syndrome) is indicated in the S1 and S2 analyses by single alleles at the two pseudoautosomal markers and the X-linked HPRT and the absence of the Y-specific products for amelogenin and SRY. The likelihood for a normal female to be homozygous for three STRs (thus indistinguishable from X chromosome monosomy) is approximately 1.5%. Once the additional markers in the MXY reflex marker set are incorporated in the analysis, the likelihood for a normal female to be homozygous for all the X-linked or pseudoautosomal STRs in the S1/S2 and MXY reactions is reduced to approximately 1 in 20,000.39 Therefore, such a result can be cautiously interpreted as suggestive of Turner syndrome with a recommendation that the results be considered in the context of other supportive findings such as cystic hygroma on ultrasound.
Data analysis
All data analyses were carried out by NYGH. Karyotype and FISH results from MSH were forwarded to NYGH using the assigned specimen codes to maintain anonymity. The PPV, NPV, overall sensitivity, and failure rate of the QF-PCR test were evaluated and compared with karyotype and FISH (where applicable).
Turnaround time
To accurately determine the turnaround time of the QF-PCR test, the time from specimen arrival to report signature was recorded for 212 specimens. The requisition accompanying the specimen was time and date stamped on arrival, and the final report was similarly time and date stamped after signature by the laboratory director. The total time in hours and minutes between specimen arrival and report signature was then calculated.
RESULTS
Retrospective validation specimens
The results of the 200 retrospective specimens are presented in Table 1. The results of these specimens were compared with the QF-PCR results previously obtained by General Lab (Barcelona, Spain), which had been confirmed with cytogenetic analysis. Comparing the results with the previous analyses, there were no false positives and a single false negative. The sensitivity (true positives/true positives + false negatives) of this analysis, therefore, was 98.9% (90/90 + 1). The PPV (true positives/true positives + false positives) was 100% (90/90 + 0) with all specimens classified as abnormal being true positives. The specificity (true negatives/true negatives + false positives) was also 100% (105/105 + 0). The NPV (true negatives/true negatives + false negatives) was 99.1% (105/105 + 1). The one specimen incorrectly classified as normal was a 45,X[7]/46,XX[23] mosaic. Four specimens failed for a failure rate of 2%.
Prospective study specimens
Patient demographics
In total, 1069 specimens were analyzed prospectively. The median maternal age was 37 years (range, 16–49 years). The median gestational age was 16 weeks, 0 days (range: 14 weeks, 6 days to 34 weeks, 0 days). As shown in Figure 2, the most common reason for referral was advanced maternal age (60.2%) followed by abnormal maternal serum biochemical screening results (34.6%) and abnormal ultrasound findings (10.3%). A small proportion of patients (8.7%) were referred for other reasons such as having a previous child affected with a chromosome abnormality or another genetic disorder or being a carrier of a balanced chromosomal translocation. It should be noted that some patients were referred for more than one indication, and therefore, the percentages in Figure 2 do not add up to 100%.

Reasons for referral for prospective study specimens. The reasons for referral were collected by the genetic counselor or geneticist at the time of study enrolment and reported on the specimen requisition. Totals do not add up to 100% because some patients had multiple reasons for referral. Positive biochemical screen, a maternal serum screening result (first trimester, second trimester, or integrated prenatal screen) with a risk > 1 in 200 for a chromosome abnormality; AMA, advanced maternal age defined as 35 years or older at the time of delivery; U/S abn, ultrasound abnormality; other: other reasons for referral, including family history of a chromosome or single gene disorder, previous child affected with a chromosome or single gene disorder, and maternal anxiety.
QF-PCR results
The QF-PCR results for the prospective study specimens are presented in Table 2. The QF-PCR results were compared with the karyotype, which was considered the gold standard. Overall, 984 of 1069 (92.1%; 483 females and 501 males) specimens had normal karyotypes. Compared with karyotyping, there were no false positives and a single false negative by QF-PCR. A total of 51 specimens (4.8%) had anomalies detectable by QF-PCR (Table 2). Of the abnormal specimens, 50 of 51 (sensitivity = 98%) were correctly identified as abnormal by QF-PCR with one mosaic sex chromosome anomaly (47,XXX[5]/46,XX[12]) classified as normal. All 50 specimens classified as abnormal were true positives to give a PPV of 100%. There were an additional 18 specimens (1.7%) with anomalies that did not include the chromosomes assayed by the QF-PCR test (see Table, Supplemental Digital Content 1, http://links.lww.com/GIM/A128, for a summary of these anomalies). Therefore, the sensitivity of the QF-PCR test to detect any abnormality was 72.5% (50/69 anomalies detected). Of the 1002 specimens categorized as negatives, 1001 were true negatives for anomalies detectable by QF-PCR (specificity, 100%; NPV, 99.9%). Four specimens (0.4%) failed, and 11 specimens (1%) had MCC that was severe enough to prevent an accurate interpretation of QF-PCR results. Finally, two specimens (0.2%) had inconclusive results with multiple normal and multiple abnormal markers for at least one chromosome. Therefore, a result was possible for 98.4% of all specimens received.
QF-PCR results were also compared with FISH results, when FISH was performed as part of regular prenatal care at the originating hospital (Table 3). FISH was performed for 251 of 1069 specimens (23.5%) that met the minimum risk criteria in place at the time of the study to determine FISH eligibility. Of these, 37 (14.7%) were abnormal and concordant between QF-PCR and FISH. Four specimens (1.6%) could not be interpreted by QF-PCR due to MCC (three specimens) or inconclusive results (one specimen). One specimen (0.4%) was a false-positive trisomy 21 by FISH but correctly interpreted as normal by QF-PCR. Therefore, the PPV was 100% (37/37) for QF-PCR and 97.4% (37/38) for FISH. The remaining 210 (83.3%) specimens were interpreted as normal by both QF-PCR and FISH, although eight (3.2%) specimens had other abnormalities detected at karyotype. The overall concordance between QF-PCR and FISH, when both assays were possible and interpretable, was 99.6% (246/247).
Turnaround time
Turnaround time was monitored for 212 specimens. The distribution of turnaround time for these specimens is presented in Figure 3. The mean turnaround time was 30.5 hours, whereas the median turnaround time was 25.1 hours. More than 90% of specimens were reported within 48 hours, and no specimen took longer than 96 hours to complete. Note that turnaround time was calculated on actual days/hours and not working days and, as such, weekend and holidays served to lengthen the calculated turnaround times.

Turnaround times from specimen receipt to report signing for 212 prospective study specimens. Requisitions were time and date stamped at the time of receipt, and reports were time and date stamped at the time of report signing. The turnaround time was calculated to the nearest minute. The majority of specimens (78.9%) were reported the day following specimen receipt. More than 90% of specimens (91.1%) were reported within 48 hours of specimen receipt. No specimen took longer than 96 hours to report.
DISCUSSION
A rapid prenatal test for aneuploidy should fulfill certain criteria: (1) highly accurate with a minimum number of false-negative results; (2) no false-positive results, as irreversible decisions such as pregnancy termination may be taken as a result of an abnormal result; (3) robust with few ambiguous results and failures; (4) rapid and capable of high specimen throughput; (5) low cost, as the rapid test is likely to be performed in addition to full karyotype analysis; (6) capable of dealing with specimens of low quality and/or quantity; and (7) detects MCC and mosaicism.9 In this study, QF-PCR met all these criteria. The analytical sensitivity and specificity were 98.9% and 100%, respectively, in a series of 200 known specimens using the commercially available Aneufast™ kit. Similarly, in a series of 1069 prospectively collected amniotic fluid specimens, the analytical sensitivity for detectable chromosome anomalies was 98%, and the specificity was 100%. The PPV was 100% in both the retrospective validation set and the prospective specimens, and the NPV was 99.1% and 99.9%, respectively.
As expected, chromosome mosaicism challenged the sensitivity of the molecular test in detecting the presence of different cell lines. In both the validation and prospective specimens, the only discrepancy was a single sex chromosome mosaic specimen classified as normal by QF-PCR. Depending on the proportion of the different cell lines, 45,X/46,XX mosaicism can be recognized by the unbalanced allelic ratios of the markers used on the X chromosome; however, at least 15–20% of the abnormal cell line should be present to sufficiently skew the ratios outside normal values.40,41 Dilution studies in our laboratory using three sets of mother and child pairs showed that a minimum of 5–10% mosaicism could be reliably detected for triallelic trisomies but that 10–20% mosaicism could be reliably detected for diallelic markers (data not shown). Similarly, mosaicism with an almost equal ratio (∼50%) of 45,X and 47,XXX cells cannot be detected by QF-PCR because they produce the same allelic patterns of normal female fetuses. It should be noted that QF-PCR and karyotype analysis are assaying two different sample types (i.e., direct specimen versus dividing culture, respectively), and, therefore, the level of mosaicism between the two tests may not always equate.
The failure rate was higher in the validation set at 2% compared with 0.4% in the prospective specimens. This was likely due to the fact that the DNA for the validation specimens had been stored at −20°C for up to 5 years before being transported from Barcelona to Toronto. Consequently, there may have been degradation of some specimens, which prevented their analysis. MCC was not an issue with the retrospective validation specimens, as these were previously tested specimens, but approximately 1% of prospective specimens had a level of MCC high enough to preclude accurate interpretation of QF-PCR results. It is possible that this issue could be overcome, or at least reduced, by proactively collecting a maternal specimen for comparison purposes when a prenatal specimen is noted to be bloody, as has been suggested by others.30 However, maternal specimens were not collected in this study. Finally, the average and median turnaround times were 30.5 and 25.1 hours, respectively, for a subset of specimens in the prospective study, with more than 90% of specimens reported within 48 hours.
In North America, the primary method used for rapid prenatal aneuploidy detection is interphase FISH. In this study, it has been demonstrated that for nonmosaic specimens, QF-PCR is equivalent to FISH in sensitivity, NPV, and timeliness of the results. The specificity and PPV of QF-PCR were superior to FISH in this study due to a false-positive trisomy 21 result by FISH. This false-positive result was most likely due to a technical error causing contamination of the DNA probe mixtures during application to the slide.42,43 Other studies have also concluded that QF-PCR is at least equivalent to interphase FISH for the prenatal detection of aneuploidy.15,27,30,34 In addition, there are several advantages of QF-PCR over interphase FISH:
-
1
Less fetal material is required to perform QF-PCR: ≤1 mL of amniotic fluid versus the 5–10 mL required for FISH.
-
2
Identification of a second cell line: mosaicism or MCC contributing to at least 15–20% of the total cell population can be confidently identified by QF-PCR. Although FISH can detect MCC in the case of a male fetus, it will not detect MCC in specimens from female fetuses.
-
3
Reduces the risk of misdiagnosis due to a small regional imbalance: although failure to detect regional imbalance can occur with QF-PCR, it is reduced with the use of multiple markers along the length of each chromosome compared with a single locus per chromosome for FISH.
-
4
Lower reagent and labor costs: QF-PCR reagents are 25% of that of FISH, and the labor costs are <50% of that of FISH. Combined, the total cost of QF-PCR is 1/3 the cost of FISH.
-
5
Potential for automation: use of semiautomated equipment (liquid handling equipment to set up PCR; automated DNA sequencer) allows for higher throughput and thus faster turnaround times. Capillary genetic analyzers are higher throughput and faster than automated FISH spot counters.
-
6
Zygosity can be determined in multiple pregnancies independently from fetal sex: the comparison of STR profiles between fetuses in multiple pregnancies allows the determination of zygosity, which may be important in the case of discordant ultrasound findings.
The availability of a rapid prenatal test for common chromosomal aberrations for all women undergoing an invasive prenatal procedure has the potential to alleviate parental anxiety and give the couple more time for decision making, which could include earlier termination of a pregnancy. Because of the high cost and relatively high labor intensity of FISH analysis, however, most centers in Ontario offer it only to those women at greatest risk. The exact criteria used to determine eligibility for FISH vary between centers, thereby creating inequity across the province. In addition, by limiting FISH testing to high-risk pregnancies, many affected pregnancies are not identified until the full karyotype analysis is available approximately 2 weeks after the amniocentesis procedure. In 2005, 1654 amniotic fluid specimens were received in the Cytogenetics Laboratory at NYGH, and karyotypes were performed for all of them. Of these, 309 specimens also had FISH analysis performed because the pregnancies met the hospital's criteria for offering FISH. Of the 1345 specimens that did not have FISH, 58 (4.3%) had a cytogenetic abnormality. That is, 4.3% of the women who were not eligible for FISH were eventually found to have a chromosomal aberration in the fetus. Of these, 34 (59% of the anomalies) would have been detected had FISH been performed.
Although both QF-PCR and FISH will detect the most common aneuploidies, these methods will not detect other clinically significant chromosomal rearrangements, such as translocations, deletions, duplications, inversions or insertions, marker chromosomes, and submicroscopic imbalances. It has been suggested, however, that QF-PCR, either alone or together with ultrasound examination, has the potential to detect at least 95% of clinically relevant chromosome anomalies.30,34 Some authors have suggested, therefore, that QF-PCR or another rapid prenatal aneuploidy test could ultimately replace karyotyping, at least in low-risk pregnancies.15,20,30,35–38 One study suggested that combining QF-PCR as a first-line test with full karyotyping only in fetuses with nuchal translucency exceeding 4 mm at 11–13 weeks of gestation would reduce the number of full chromosome analyses required by approximately 90% while detecting 99% of clinically relevant anomalies and costing 60% less than full karyotyping for every pregnancy.35
In this study, 27.9% of chromosome anomalies were not detected by QF-PCR. These included a mosaic case (47,XXX[5]/46,XX[12] not detected by the molecular assay) and 18 other anomalies that would not be detected by the QF-PCR assay. These included translocations, inversions, deletions, marker chromosomes, and mosaicism for aneuploidy for a chromosome not included in the QF-PCR kit. Of these 18, 10 (55.6%) were balanced translocations or inversions inherited from a phenotypically normal carrier parent. An additional two (11.1%) were apparently balanced translocations or inversions that were de novo in the fetus. If balanced rearrangements are interpreted as having a low risk of adverse outcomes in the fetus, the QF-PCR assay detected 50 of 57 (87.7%) of clinically relevant chromosome anomalies (i.e., those likely to result in a genomic imbalance and a high risk of adverse outcomes in the fetus) in this series of patients. It is possible that incorporating other risk factors such as abnormal ultrasound findings would identify those pregnancies likely to be affected with a chromosome anomaly other than those detected by the QF-PCR assay; however, this analysis was not done in this study.
The results of this study compare favorably with other large studies of the use of QF-PCR for aneuploidy in fetal specimens, which also demonstrated a high level of concordance with karyotype results and detection of the majority of clinically significant chromosome anomalies.16,21,28,30
This study has several strengths. First, it was prospective in nature, with all women undergoing amniocentesis eligible for the study and specimens analyzed in real time as they were received in the laboratory. Second, the assay was validated on a large number of previously characterized normal and abnormal specimens before use in the prospective study. Third, both the validation and prospective studies were conducted in a blind fashion, so that interpretation was not influenced by knowledge of the expected outcome. Finally, more than 1000 specimens were analyzed prospectively, such that all commonly expected abnormal results were observed among the study specimens and some unusual results (e.g., 49,XXXXY), thereby providing valuable experience in the interpretation of QF-PCR results.
This study also has some limitations. The study participants were self-selected, and because no analysis of outcomes in women who did not participate was performed, it is not clear whether the study population is representative of all women undergoing amniocentesis at NYGH or MSH. However, because approximately 60% of the women did elect to participate, it is expected that the results are fairly representative of the population. In addition, although the study was conducted in real time with specimens being processed the same day they arrived in the laboratory, turnaround time was only evaluated in approximately 20% of specimens. These specimens were unselected and represented the final 20% of specimens analyzed in the study. However, having offered QF-PCR as service for the past 2.5 years at NYGH, where the majority of the samples (>99%) are reported the next working day, the turnaround times of the study seem to have been representative. This is comparable with FISH turnaround times, which averaged 1.3 days during the 2 final years that FISH was offered at NYGH. Finally, this study was limited to amniotic fluid specimens. Other studies have reported the successful use of QF-PCR with CVS specimens, however, and thus, it is expected that the technology could easily be validated for CVS.21,30
This is the largest study to date in North America that prospectively evaluated the performance of QF-PCR in the prenatal diagnosis of aneuploidy for chromosomes 13, 18, 21, X, and Y. The results of this study show that QF-PCR is a viable alternative to interphase FISH as a rapid prenatal diagnostic test for common aneuploidies. Further research is needed to determine whether QF-PCR could replace full karyotyping in low-risk pregnancies in the context of the Canadian health care system. It should be noted, however, that a recent study in the United Kingdom found that using QF-PCR as a standalone test for pregnancies without ultrasound anomalies resulted in faster turnaround time, lower costs, and avoidance of ambiguous karyotype results.21
REFERENCES
Chodirker B, Cadrin C, Davies G, et al. Canadian guidelines for prenatal diagnosis: genetic indications for prenatal diagnosis. J Soc Obstet Gynaecol Can 2001; 23: 525–531.
Summers AM, Langlois S, Wyatt P, Wilson RD . Prenatal screening for fetal aneuploidy. J Obstet Gynaecol Can 2007; 29: 146–179.
Hassold T, Hunt PA, Sherman S . Trisomy in humans: incidence, origin and etiology. Curr Opin Genet Dev 1993; 3: 398–403.
Homer J, Bhatt S, Huang B, Thangavelu M . Residual risk for cytogenetic abnormalities after prenatal diagnosis by interphase fluorescence in situ hybridization (FISH). Prenat Diagn 2003; 23: 566–571.
Wilson RD, Davies G, Gagnon A, et al. Amended Canadian guideline for prenatal diagnosis (2005) change to 2005-techniques for prenatal diagnosis. J Obstet Gynaecol Can 2005; 27: 1048–1062.
Test and Technology Transfer Committee. Technical and clinical assessment of fluorescence in situ hybridization: an ACMG/ASHG position statement. I. Technical considerations. Genet Med 2000; 2: 356–361.
Tepperberg J, Pettenati MJ, Rao PN, et al. Prenatal diagnosis using interphase fluorescence in situ hybridization (FISH): 2-year multi-center retrospective study and review of the literature. Prenat Diagn 2001; 21: 293–301.
Shaffer LG, Bui T . Molecular cytogenetic and rapid aneuploidy detection methods in prenatal diagnosis. Am J Med Genet C Semin Med Genet 2007; 145C: 87–98.
Mann K, Donaghue C, Fox SP, Docherty Z, Ogilvie CM . Strategies for the rapid prenatal diagnosis of chromosome aneuploidy. Eur J Hum Genet 2004; 12: 907–915.
Diego-Alvarez D, Rodriguez de Alba M, Cardero-Merlo R, et al. MLPA as a screening method of aneuploidy and unbalanced chromosomal rearrangements in spontaneous miscarriages. Prenat Diagn 2007; 27: 765–771.
Fernández-Martínez FJ, Galindo A, Moreno-Izquierdo A, et al. Application of QF-PCR for the prenatal assessment of discordant monozygotic twins for fetal sex. Prenat Diagn 2007; 27: 648–652.
Diego-Alvarez D, Garcia-Hoyos M, Trujillo MJ, et al. Application of quantitative fluorescent PCR with short tandem repeat markers to the study of aneuploidies in spontaneous miscarriages. Hum Reprod 2005; 20: 1235–1243.
Cirigliano V, Lewin P, Szpiro-Tapies S, Fuster C, Adinolfi M . Assessment of new markers for the rapid detection of aneuploidies by quantitative fluorescent PCR (QF-PCR). Ann Hum Genet 2001; 65: 421–427.
Cirigliano V, Ejarque M, Cañadas MP, et al. Clinical application of multiplex quantitative fluorescent polymerase chain reaction (QF-PCR) for the rapid prenatal detection of common chromosome aneuploidies. Mol Hum Reprod 2001; 7: 1001–1006.
Schmidt W, Jenderny J, Hecher K, et al. Detection of aneuploidy in chromosomes X, Y, 13, 18 and 21 by QF-PCR in 662 selected pregnancies at risk. Mol Hum Reprod 2000; 6: 855–860.
Mann K, Fox SP, Abbs SJ, et al. Development and implementation of a new rapid aneuploidy diagnostic service within the UK National Health Service and implications for the future of prenatal diagnosis. Lancet 2001; 358: 1057–1061.
Andonova S, Vazharova R, Dimitrova V, et al. Introduction of the QF-PCR analysis for the purposes of prenatal diagnosis in Bulgaria—estimation of applicability of 6 STR markers on chromosomes 21 and 18. Prenat Diagn 2004; 24: 202–208.
Mann K, Petek E, Pertl B . Prenatal detection of chromosome aneuploidy by quantitative fluorescence PCR. Methods Mol Biol 2008; 444: 71–94.
El Mouatassim S, Becker M, Kuzio S, et al. Prenatal diagnosis of common aneuploidies using multiplex quantitative fluorescent polymerase chain reaction. Fetal Diagn Ther 2004; 19: 496–503.
Bili C, Divane A, Apessos A, et al. Prenatal diagnosis of common aneuploidies using quantitative fluorescent PCR. Prenat Diagn 2002; 22: 360–365.
Hills A, Donaghue C, Waters J, et al. QF-PCR as a stand-alone test for prenatal samples: the first 2 years' experience in the London region. Prenat Diagn 2010; 30: 509–517.
Lampret J, Lane T, Christianson A . QF-PCR for prenatal diagnosis of common aneuploides in women of advanced maternal age. S Afr Med J 2008; 98: 68–69.
Quaife R, Wong LF, Tan SY, et al. QF-PCR-based prenatal detection of aneuploidy in a southeast Asian population. Prenat Diagn 2004; 24: 407–413.
Putzova M, Soldatova I, Pecnova L, et al. QF-PCR-based prenatal detection of common aneuploidies in the Czech population: five years of experience. Eur J Med Genet 2008; 51: 209–218.
Adinolfi M, Pertl B, Sherlock J . Rapid detection of aneuploidies by microsatellite and the quantitative fluorescent polymerase chain reaction. Prenat Diagn 1997; 17: 1299–1311.
Pertl B, Pieber D, Lercher-Hartlieb A, et al. Rapid prenatal diagnosis of aneuploidy by quantitative fluorescent PCR on fetal samples from mothers at high risk for chromosome disorders. Mol Hum Reprod 1999; 5: 1176–1179.
Ochshorn Y, Bar-Shira A, Jonish A, Yaron Y . Rapid prenatal diagnosis of aneuploidy for chromosomes 21, 18, 13, and X by quantitative fluorescence polymerase chain reaction. Fetal Diagn Ther 2006; 21: 326–331.
Ogilvie CM, Donaghue C, Fox SP, Docherty Z, Mann K . Rapid prenatal diagnosis of aneuploidy using quantitative fluorescence-PCR (QF-PCR). J Histochem Cytochem 2005; 53: 285–288.
Onay H, Ugurlu T, Aykut A, et al. Rapid prenatal diagnosis of common aneuploidies in amniotic fluid using quantitative fluorescent polymerase chain reaction. Gynecol Obstet Invest 2008; 66: 104–110.
Cirigliano V, Voglino G, Ordoñez E, et al. Rapid prenatal diagnosis of common chromosome aneuploidies by QF-PCR, results of 9 years of clinical experience. Prenat Diagn 2009; 29: 40–49.
Cirigliano V, Voglino G, Cañadas MP, et al. Rapid prenatal diagnosis of common chromosome aneuploidies by QF-PCR. Assessment on 18,000 consecutive clinical samples. Mol Hum Reprod 2004; 10: 839–846.
Liao C, Yang X, Li F, Li J, Li D . The detection of aneuploidy and maternal contamination by QF-PCR in samples undergoing prenatal diagnosis for thalassemia in Southern China. Eur J Obstet Gynecol Reprod Biol 2009; 144: 149–152.
Cho EH, Park BYN, Kang YS, Lee EH . Validation of QF-PCR in a Korean population. Prenat Diagn 2009; 29: 213–216.
Brown L, Abigania M, Warburton D, Brown S . Validation of QF-PCR for prenatal aneuploidy screening in the United States. Prenat Diagn 2006; 26: 1068–1074.
Chitty LS, Kagan KO, Molina FS, Waters JJ, Nicolaides KH . Fetal nuchal translucency scan and early prenatal diagnosis of chromosomal abnormalities by rapid aneuploidy screening: observational study. BMJ 2006; 332: 452–455.
Speevak MD, Dolling J, Terespolsky D, Blumenthal A, Farrell SA . An algorithm for the prenatal detection of chromosome anomalies by QF-PCR and G-banded analysis. Prenat Diagn 2008; 28: 1221–1226.
Gaudry P, Lebbar A, Choiset A, et al. Is rapid aneuploidy screening used alone acceptable in prenatal diagnosis? An evaluation of the possible role of ultrasound examination. Fetal Diagn Ther 2009; 25: 285–290.
Sparkes RL, Bernier FP, Chernos JE, Johnson JM . Suitability of rapid aneuploidy detection for prenatal diagnosis. J Obstet Gynaecol Can 2008; 30: 781–787.
molGENTIX, S.L. Aneufast User Manual, 2007. Available at: http://www.aneufast.com/files/ANEUFASTUsersManual%20July%202007%20nm.pdf. Accessed July 22, 2010.
Donaghue C, Mann K, Docherty Z, Ogilvie CM . Detection of mosaicism for primary trisomies in prenatal samples by QF-PCR and karyotype analysis. Prenat Diagn 2005; 25: 65–72.
Cirigliano V, Sherlock J, Conway G, et al. Rapid detection of chromosomes X and Y aneuploidies by quantitative fluorescent PCR. Prenat Diagn 1999; 19: 1099–1103.
Sroka H, Kolomietz E, Winsor E, et al. Prenatal false positive for trisomy 21 with fluorescent in situ hybridization (FISH). In: Abstract 2404. American Society of Human Genetics Annual Meeting, San Diego, CA, 2007.
Wang J, Bowser K, Chernos J . Shedding new light on false positive diagnosis of trisomy 21 by fluorescence in situ hybridization (FISH) on uncultured amniotic fluid cells: experiences from two Canadian cytogenetic laboratories. Prenat Diagn 2007; 27: 964–966.
Acknowledgements
This study was supported by research Grants from the Canadian Foundation for Innovation (CFI) Canadian Molecular Cytogenetics Platform (Grant no. 8689) and the Ontario Association of Medical Laboratories (OAML). The authors thank the technical staff in the Molecular Genetics and Cytogenetics laboratories at NYGH for technical assistance; the technical staff in the Cytogenetics laboratory at MSH for preparing and shipping the specimens from MSH to the NYGH laboratory; and the geneticists, obstetricians, and genetic counselors at NYGH and MSH for enrolling their patients in the study. Most importantly, the authors thank the patients who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
Allingham-Hawkins, D., Chitayat, D., Cirigliano, V. et al. Prospective validation of quantitative fluorescent polymerase chain reaction for rapid detection of common aneuploidies. Genet Med 13, 140–147 (2011). https://doi.org/10.1097/GIM.0b013e3182036763
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3182036763
Keywords
This article is cited by
-
QF-PCR: a valuable first-line prenatal and postnatal test for common aneuploidies in South Africa
Journal of Community Genetics (2022)
-
Medical Genetics for Practicing Obstetrician
The Journal of Obstetrics and Gynecology of India (2020)
