
Abstract
Purpose
Blood/saliva DNA is thought to represent the germ line in genetic cancer-risk assessment. Cases with pathogenic TP53 variants detected by multigene panel testing are often discordant with Li-Fraumeni syndrome, raising concern about misinterpretation of acquired aberrant clonal expansions (ACEs) with TP53 variants as germ-line results.
Methods
Pathogenic TP53 variants with abnormal next-generation sequencing metrics (e.g., decreased ratio (<25%) of mutant to wild-type allele, more than two detected alleles) were selected from a CLIA laboratory testing cohort. Alternate tissues and/or close relatives were tested to distinguish between ACE and germ-line status. Clinical data and Li-Fraumeni syndrome testing criteria were examined.
Results
Among 114,630 multigene panel tests and 1,454 TP53 gene-specific analyses, abnormal next-generation sequencing metrics were observed in 20% of 353 TP53-positive results, and ACE was confirmed for 91% of cases with ancillary materials, most of these due to clonal hematopoiesis. Only four met Chompret criteria. Individuals with ACE were older (50 years vs. 33.7; P = 0.02) and were identified more frequently in multigene panel tests (66/285; 23.2%) than in TP53 gene-specific tests (6/68; 8.8%, P = 0.005).
Conclusion
ACE confounds germ-line diagnosis, may portend hematologic malignancy, and may provoke unwarranted clinical interventions. Ancillary testing to confirm germ-line status should precede Li-Fraumeni syndrome management.
Similar content being viewed by others

Introduction
DNA isolated from peripheral blood or saliva is typically considered representative of the germ line for diagnosis of hereditary cancer. Since the advent of next-generation sequencing (NGS), multigene panel testing (MGPT) for hereditary cancer has been increasingly utilized in genetic cancer risk assessment.1 MGPT is a potential cost- and time-effective alternative to sequential gene testing, yet clinicians may find themselves dealing with unexpected findings, such as the detection of a cancer gene variant that is not known to correlate with the cancers in the patient and/or his or her family.2,3 Li-Fraumeni syndrome (LFS) was initially described by Frederick Li and Joseph Fraumeni in 19694 and associated with TP53 in 1990.5 The most frequently occurring tumors recognized as core cancers in LFS are sarcomas, breast cancers, central nervous system tumors, and adrenocortical carcinomas.6,7,8,9
Various groups have published criteria to define LFS and identify patients to undergo TP53 gene-specific testing.10,11,12,13,14,15 Given the broad tumor spectrum described in LFS families, TP53 is included in most cancer-focused MGPT, and has resulted in the identification of carriers with a phenotype that is discordant with what has been reported for LFS,16 suggesting the possibility of an expanded phenotype associated with germ-line TP53 variants. However, the observation of a decreased mutant to wild-type allele ratio for a substantial portion of TP53 variants detected through NGS raises the question of whether clonal populations in the blood or saliva that do not represent the germ line or early postzygotic mosaicism were confounding clinical diagnosis. In most cases, a type of postzygotic variation described by Forsberg et al. as aberrant clonal expansions (ACE) is suspected.17 We previously applied the term “somatic interference” to describe a circumstance wherein the analytical results of the test are technically valid (a pathogenic TP53 variant was detected) yet the intent was to identify a bona fide germ-line predisposition to cancer.18 However, for the purposes of this paper, we refer to this phenomenon as ACE.
The term “ACE” is used in distinction to the relatively infrequently documented phenomenon of classic mosaicism involving TP53, wherein a variant is acquired during embryogenesis and is variably present in one or multiple germ layers, conferring increased cancer risk in the respective tissues as well as the possible transmission of risk to offspring.19,20 True somatic mosaicism has been well-documented for NF1 and NF2 cancer-associated genes and there has been one documented case for PTEN and Cowden syndrome.21,22,23,24 There is a growing literature documenting the detection of mosaic mutations in disease genes detected by NGS, albeit in the context of developmental disorders manifesting in childhood.25 However, in this study our focus is on the possibility that late postzygotic ACE, limited to the hematologic compartment or to a tumor, may be detected in the blood or saliva in the context of NGS-based testing to detect germ-line cancer predisposition. The magnitude of this phenomenon is yet unknown, and its potential impact on clinical care must be considered, given that more than 50,000 MGPTs are conducted every year across a growing number of commercial vendors, and many clinicians have limited experience of ordering and interpreting MGPT.1,26 Further, there is increasing evidence of the effectiveness of surveillance regimens prescribed for individuals with a germ-line TP53 variant.27,28 However, these surveillance measures are resource-intensive and have the potential for adverse events, so it is important to apply them only to individuals truly at risk.
This study evaluated the prevalence and possible causes of apparent ACE involving TP53 in a large series of patients who had clinical MGPT or TP53 gene-specific testing.
Materials and methods
Study population
A system-wide search of the Ambry Genetics (Aliso Viejo, CA) Laboratory Information Management System for clinical cases tested with MGPT that included TP53 or NGS-based TP53 gene-specific tests between March 2013 and February 2016 was conducted. Cases were selected with test results reporting TP53 pathogenic and likely pathogenic variants (TP53 variants) with abnormal NGS metrics, including (i) a minor allele frequency (MAF) of less than 25% or (ii) an MAF between 25–30% if the clinical history or molecular results were suspicious for ACE (e.g., LFS criteria not met, active hematologic malignancy, or multiple mutations detected) and Sanger results were consistent with the MAF. Multiple or atypical abnormalities detected on microarray looking at large rearrangements were also included.
All TP53 variants detected by NGS were confirmed on Sanger sequencing; gross deletions/duplications were evaluated using multiplex ligation-dependent probe amplification and/or microarray.
Data collection
Demographic, personal, and family history information was collected from test requisition forms (patient gender, age at testing, cancer type, age at cancer diagnosis, ER/PR/HER2 receptor status for breast cancer, and a family cancer history table), clinic notes, pedigrees, letters of medical necessity, and medical records submitted to Ambry Genetics. Additional information, such as personal history of hematologic neoplasia, extended cancer family history (first- to third-degree relatives), and other genetic test results, was collected through direct communication with the individuals’ health-care providers. The study was approved by Solutions IRB and the City of Hope Institutional Review Board. Case-specific details have been amended to obscure characteristics by which individuals might be identified.
Laboratory methods
Multigene panel tests and TP53 gene-specific testing
Both MGPT and TP53 gene-specific tests were performed using DNA isolated from whole blood or saliva samples. NGS analysis (Illumina, San Diego, CA) was performed in all coding domains plus at least five bases into the 5′ and 3′ ends of the introns and untranslated regions (5′ UTR and 3′ UTR) for all cancer susceptibility genes. EPCAM and GREM1, if included on the panel, were analyzed only for gross deletions and duplications. Depending on the panel ordered by the clinician, 5–49 genes, including TP53, were analyzed. Sanger sequencing was performed for any region with insufficient depth of coverage (<10 ×), for verification of all variants (other than known benign variants), and for those with decreased mutant to wild-type allele ratios. A targeted chromosomal microarray and/or multiplex ligation-dependent probe amplification was used for the detection of gross deletions and duplications.
A five-tier classification schema—pathogenic; variant, likely pathogenic; variant of unknown significance; variant, likely benign; and benign—was used to classify variants.29
Single-site analyses of ancillary tissues for known TP53 variant(s)
Single-site analysis for previously identified TP53 variants was performed on available tissue samples. DNA was extracted from formalin-fixed, paraffin-embedded tissue, fibroblasts cultured from skin, and eyebrow plucks.30 Single-site analysis was conducted using Sanger sequencing for all cases; in certain cases NGS was also performed.
Single-site analysis in family members
Testing for transmission of the known TP53 variant was offered to first-degree relatives. When two TP53 variants were detected, Sanger sequencing was performed for both.
Data analyses
Using descriptive statistics, exact binomial confidence limits were calculated at 95%. Tests of difference between more than two groups for binary variables use Fisher’s exact test to generate two-tailed P values. Student’s t-test was used to compare the means of ages at diagnosis and years since diagnosis. The clinical features of each case were assessed against the published criteria for TP53 testing (National Comprehensive Cancer Network, Chompret, classic) (Supplementary Table S1 online).9,10,31,32
Data sharing
Ambry genetic testing data are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and access can also be obtained via application to the AmbryShare program (https://share.ambrygen.com/about-ambry-share) (Ambry Genetics, Aliso Viejo, CA).
Results
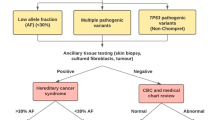
Among 114,630 MGPTs that included TP53 and 1,454 gene-specific TP53 analyses, 353 cases were identified with a TP53 variant (representing only pathogenic or likely pathogenic variants, as noted in Materials and Methods). Seventy-two cases (20.4%) were selected with test results reporting TP53 pathogenic and likely pathogenic variants (TP53 variants) with abnormal NGS metrics, and these cases were selected for further study (study cohort). Sixty-six patients had an MAF less than 25%, three had an MAF between 25 and 30%, with a clinical history and molecular results suspicious for ACE and Sanger results consistent with the MAF, and three had multiple or atypical abnormalities on microarray looking at large rearrangements. All cases were reviewed by a laboratory director before a clinical report was issued (Figure 1, Table 1). The suspected ACE cases represented a significantly higher proportion (66/285; 23.2%) of MGPT cases than of those undergoing TP53 gene-specific testing (6/68; 8.8%, P = 0.005). The mean age at testing was significantly older (58.4 vs. 44.3 years; P < 0.0001) for suspected ACE cases than it was for those with unambiguous NGS metrics among the MGPT cases; a similar pattern was observed among the six TP53 gene-specific cases (39.5 vs. 30.1 years).

Consort diagram of the clinical study. The total sample of cases evaluated at the genetic testing laboratory is indicated, followed by the subsets of MGPT and single-gene TP53 tests and the subsets meeting eligibility criteria. Cases that have and have not undergone ancillary testing are noted, as is the final assignment of ACE status, based on consideration of the data. ACE, aberrant clonal expansion; MGPT, multigene panel test; NGS, next-generation sequencing; P, pathogenic; VLP, variant likely pathogenic.
The study cohort was predominantly female (95.8%), with a personal history of cancer in 68 of 72 individuals (94.4%). Figure 2a depicts the types and prevalence of cancers observed in the study cohort. Eighteen cases had multiple primary cancers. The average age at onset for first cancer was 48.2 years (2–80 years). The average age at the time of genetic testing was 57 years (15–86 years) (Table 1).

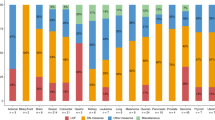
Spectrum of cancers among cases and their relatives (a) Spectrum of cancers among TP53 cases suspected to be due to ACE. The cancer subtypes and their respective proportions are indicated in the pie chart. Note that 18 individuals had multiple tumor types. (b) Spectrum of cancers among relatives of cases suspected to be due to ACE; The cancer subtypes (excluding nonmelanoma skin cancers) reported among first- and second-degree relatives, and their respective proportions, are indicated in the pie chart. aUrothelial and kidney: bladder, kidney, renal pelvis. bHead and neck: esophageal, laryngeal, nose, throat. MDS, myelodysplastic syndrome.
Ninety-two cancers were reported among the 68 affected individuals (Figure 2a). Breast cancer was the most common diagnosis (53%), with an average age at diagnosis of 46 years (19–72 years). Seven cases had a breast cancer diagnosis under the age of 31 years (National Comprehensive Cancer Network criterion).32 The status of the hormone receptors (estrogen and progesterone) and HER2 amplification was available for 25 breast cancer cases; of which 18 were estrogen receptor–positive (72%), two were triple-positive (8%), and six were triple-negative (24%).
Ovarian cancer (n = 19; 27.9%) was the second most common diagnosis, with an average age at diagnosis of 60.1 years (43–74 years) (Figure 2a). The average time between ovarian cancer diagnosis and genetic testing was four years (0–12 years). Seven (37%) of these cases had at least one additional primary cancer.
Fifty-seven individuals reported a family history of cancer among first- and/or second-degree relatives (Figure 2b). Two reported a relative with childhood cancer (diagnosis at <18 years of age). Breast, colorectal, prostate, and uterine cancers were the most frequently reported among relatives. Two reported a relative with sarcoma (soft tissue or bone), five a relative with a central nervous system tumor, and one a relative with an adrenocortical carcinoma. Four of 72 (5.6%) cases met Chompret or classic diagnostic LFS criteria.
The majority (91.6%) of cases suspicious for ACE were identified through MGPT. Of these, four (6.1%) had two distinct TP53 variants, both with a low MAF. Eight individuals (12.1%) also had a pathogenic variant in another cancer-predisposition gene (Table 2), including one in PMS2 with a low MAF. The other seven (ATM, BRCA2, BRIP1, MLH1, MUTYH, PALB2, RAD50) appeared to be germ-line findings, among which four had clinical phenotypes compatible with the respective gene.
Five of seven cases with a history of hematological neoplasia were determined to have evidence of active disease at the time of testing: three with myelodysplastic syndrome, one with chronic lymphocytic leukemia, and one with acute myeloid leukemia.
Ancillary testing, consisting of additional tissue analyses and/or single-site analysis of relatives, was performed on 35 of 72 cases (48.6%) (Figure 1). Twenty-two cases had a total of 62 relatives undergo single-site analysis. Germ-line status was confirmed in 3 of the 35 (8.6%) cases with additional testing: the respective TP53 variant was detected in at least one family member for two of the TP53 gene-specific testing cases. One was a 34-year-old woman with breast cancer whose twin sister (zygosity uncertain) was also found to carry the TP53 variant. The other was a 15-year-old boy with acute leukemia and a history of rhabdomyosarcoma and myelodysplastic syndrome in early childhood; he had two TP53 variants (one missense variant and one large deletion). Only the missense TP53 variant was detected in his mother, who had a neuroendocrine tumor, and in the offspring of his maternal aunt, who was diagnosed with osteosarcoma at age 16 and a brain tumor at age 25. Single-site analyses of family members for all the other cases (n = 20) were negative for transmission of the respective TP53 variants. The third case, a 19-year-old woman with triple-negative breast cancer, was one of the MGPT cases, and the respective TP53 variant was detected in cultured skin fibroblasts.
Additional tissue was collected from 19 individuals, of whom seven had more than one unique tissue type available for analysis. The respective TP53 variant was not detected in 27 nonlymphoid tissue samples, supporting a conclusion of ACE for the original sample submitted for clinical germ-line testing. Of the cases in which the tumor tissue was tested, none appeared to be the origin of the TP53 variant detected on germ-line testing. In one illustrative case, a 79-year-old unaffected man underwent MGPT because his brother had died of pancreatic cancer. The test detected a TP53 variant (MAF = 12.7%). Subsequent testing of eyebrow plucks and cultured skin fibroblasts did not reveal the respective TP53 variant. One year after MGPT, his prostate-specific antigen level had risen to 16 and a biopsy confirmed Gleason grade 7–8 prostate cancer. The results of sequencing of the tumor tissue were negative for the variant. However, review of the aligned sequence data revealed that the variant was present in 1% of more than 10,000 reads—below the threshold for validation, but consistent with reported inflammatory cells in the biopsy specimen. These findings support a conclusion of ACE.
Analysis of benign tissue of lymphoid origin in two cases identified the respective TP53 variant. One was a 38-year-old woman with a diagnosis of splenic angiosarcoma at age 38. The TP53 variant was detected at a low level (MAF = 17%) in benign splenic tissue adjacent to the angiosarcoma. This MAF was commensurate with the MAF seen in the blood (18%). The other case was a 66-year-old woman diagnosed with breast cancer at age 45 and rectal carcinoid and lung cancer at age 56. The variant was not detected in benign duodenal and stomach tissues. It was detected in a benign lymph node and in benign colon tissue with prominent lymphocytic infiltrate; the MAF for the variant was lower in the colon tissue than in the blood (9% vs. 22%). Germ-line testing was negative for two daughters. These findings support a conclusion of ACE for both cases.
Twenty-nine of 30 MGPT cases with ancillary testing (96.7%) supported a conclusion of ACE (Table 1,Figure 1). Comparison of these cases to the remaining suspected ACE MGPT cases with no ancillary testing (n = 36) showed no significant difference in age at diagnosis of breast cancer (mean = 43.7 (25–72) years vs. mean = 49.1 (31–69) years, respectively) (P = 0.12), age at genetic testing (P = 0.2), and time between first cancer diagnosis and genetic testing (P = 0.31).
Discussion
TP53 variants are increasingly detected on MGPT across diverse patient scenarios.3,16,33,34,35,36 Although these findings may suggest a broader phenotype than is typically associated with LFS, we have demonstrated that ACE in germ-line testing is a clinically important phenomenon, involving nearly a quarter of MGPT wherein TP53 variants were detected in blood or saliva. A recent short report from another commercial diagnostic laboratory reported that 38.8% of MGPT-detected TP53 variants had abnormal NGS germ-line metrics, although ancillary testing was not performed.37 We observed a similar proportion of suspect TP53 results and evaluated ancillary tissues, providing evidence supporting the conclusion of ACE in most cases. Further, the criteria defining abnormal NGS metrics are not uniform among commercial genetic-testing laboratories, so the true prevalence of the phenomenon is uncertain and the inclusion of qualifications on the report to alert clinicians is inconsistent. Nonetheless, ancillary studies performed on appropriate tissues can eliminate consideration of specific germ layers (e.g., epithelial by virtue of a negative result in skin biopsy or eyebrow pluck), although ultimately they cannot prove that postzygotic mosaicism does not exist.
In most cases, the ACE is probably due to clonal hematopoiesis of indeterminate potential (CHIP), which can be demonstrated in healthy populations at increasing frequency with increasing age.33,34,38 Previous studies of CHIP have demonstrated increased risk (approximately 1%/year) for the development of overt hematologic neoplasia and increased overall mortality, especially if there is a variant in more than one gene involved in hematologic neoplasias (e.g., ASXL1 or DNMT3A).33,38 However, with the exception of TP53 and ATM, genes that are frequently mutated in clonal hematopoiesis are not included in most hereditary cancer MGPT.
Only 7.2% (5/69) of cases in our series that were deemed unlikely to be germ-line had evidence of overt hematological neoplasia as a likely cause of ACE (Supplementary Table S2), and only one of these was noted on the test requisition form (the others were identified by queries related to this study). Therefore, there should be clear instructions from genetic-testing laboratories regarding the unsuitability of blood and/or saliva as a source of DNA for germ-line testing in cases with a history of hematologic abnormalities. Further, careful examination of the patient’s complete blood count and peripheral smear may be warranted in all cases in which the discovery of a TP53 variant is reported.
Thirty-five of the 72 (48.6%) cases in this study had ancillary materials and/or clinical data to interrogate germ-line status, and there were no significant differences in age at breast cancer diagnosis, age at testing, and time from diagnosis to testing for those with and without ancillary data. The latter were labeled “indeterminate” in our analysis (Supplementary Table S2). However, assuming no bias in access to ancillary data or tissues, and given the apparent lack of differentiating clinical features between those with and without ancillary material and the fact that none of the cases had a clinical phenotype of LFS, we speculate that ACE probably explains the majority of the remaining cases. Nonetheless, testing of ancillary tissues would be necessary to confirm or disprove germ-line status.
Because clinical information for a proportion of the cases was limited to that available on the clinician-completed test-requisition form, supplemental efforts to obtain comprehensive personal medical and family-history data were made for all cases and ancillary tissues were obtained when possible. Follow-up e-mails and phone calls to ordering providers with requests for additional information were moderately successful, but standardized family-history collection was not uniform across the series.
Historically, when a patient’s blood or saliva has not been the appropriate specimen for germ-line genetic testing owing to a history of hematologic neoplasia or allogenic bone marrow/stem cell transplant, skin fibroblasts have been utilized for testing. However, coordination of skin-punch biopsy and cell culturing can be costly and challenging. One innovative approach employed in this study was the use of eyebrow plucks as a surrogate for skin biopsies and the growth of fibroblasts. We documented a yield of 0.5–1.2 μg of high-quality DNA from 10–15 eyebrow hair follicles. A technical manuscript outlining the specifications and standard operating procedure for processing eyebrow hair follicles to obtain genomic DNA is in process.30
In addition to cultured skin fibroblasts, alternatives to white blood cells from blood or saliva may be considered for interrogation of the germ line. One can use normal solid tissues or solid tumor tissue derived from archival surgical specimens. The possibility that circulating tumor cells could explain ACE is suggested by detection of tumor-related variants in cell-free DNA (“liquid biopsy”).39 However, none of the cases with ancillary testing of tumor tissue in our series showed the respective TP53 variant. If the variant is found in the tumor, however, one cannot determine whether it represents ACE (as a result of circulating tumor DNA) or a germ-line finding. Distinguishing germ-line from somatic variants in tumor tissues is another emerging challenge for clinicians. Finally, orthogonal testing with different methods (e.g., NGS and Sanger sequencing on the same sample) provides evidence that the skewed MAF is probably not due to a technical problem with differential allelic amplification.
Normal (noncancer) tissues are preferred for confirmatory studies, and tissues in the lymphoid compartment or with inflammatory cell infiltrate should be avoided, as they may reflect the clonal hematopoietic findings from the blood. For example, two cases of ACE in our series demonstrated the variant in benign tissue with heavy representation of lymphocytes: lymph nodes, spleen, and colon tissue with predominant inflammatory cell infiltrate. The spleen and lymph nodes are arguably part of the hematopoietic compartment. The MAF of the TP53 variant in the lymphoid-derived tissue in each of the cases in our series approximated that observed in the blood. Thus, we recommend the avoidance of ancillary tissues from the hematopoietic compartment, such as bone marrow, lymph nodes, spleen, or tissues identified through histologic analysis as having significant lymphocytic infiltrates.
Apparent CHIP was especially prevalent among the ovarian cancers in our series. The TP53 variant was not detected in any of the ancillary tissues analyzed in five ovarian cancer cases. Using NGS to analyze a set of cancer genes in the peripheral blood of women with ovarian cancer, Swisher et al.36 demonstrated that somatic mosaic variants in PPM1D were associated with chemotherapy exposure and more advanced age at the time of blood draw. Ruark et al.40 speculated about mosaicism in lymphocytes and concluded that PPM1D mutations predisposed to breast and/or ovarian cancer, although the authors were not able to detect the PPM1D mutations in any of the respective tumors; thus we believe that the more likely explanation is clonal hematopoiesis. Swisher et al.36 further observed the emergence of pathogenic TP53 variants in the blood after exposure to chemotherapy. With approximately 4 years between ovarian cancer diagnosis and subsequent genetic testing in our series, interval-chemotherapy exposure is a common feature of cases with apparent ACE. There are many potential causes of ACE, including circulating tumor DNA, but the only evidence in this case series was CHIP, or an evolving hematologic neoplasia.
Using a large clinical series we demonstrated that the phenomenon of ACE was common and was most often due to clonal hematopoiesis. This finding has important clinical implications regarding the potential application of unwarranted clinical interventions. Further, the discovery of clonal hematopoiesis itself may portend adverse clinical outcomes, such as the development of hematologic neoplasia and increased nonhematologic mortality.
Currently, there are no standard guidelines for NGS quality-control measures for detecting or reporting potential ACE. Laboratories need to be transparent about their policies regarding the detection, reporting, and follow-up of cases with potential ACE.
Confirming the validity of germ-line TP53 test results may be necessary to evaluate the associated phenotype(s) and enable accurate identification and management of germ-line carriers. Ancillary tissues should be obtained and tested to determine whether a given variant is present in any tissue other than the blood. Beyond the use of NGS quality-control measures, a clinician’s recognition of test results inconsistent with an LFS phenotype should raise the index of suspicion, and caution is urged in the medical management of patients for whom the only criterion for LFS is a TP53 variant.
References
Weitzel JN, Blazer KR, MacDonald DJ, Culver JO, Offit K. Genetics, genomics and cancer risk assessment: state of the art and future directions in the era of personalized medicine. CA Cancer J Clin 2011;61:327–359.
Blazer KR, Nehoray B, Solomon I et al. Next-generation testing for cancer risk: perceptions, experiences, and needs among early adopters in community healthcare settings. Genet Test Mol Biomarkers 2015;19:657–665.
Slavin T, Niell-Swiller M, Solomon I et al. Clinical application of multigene panels: challenges of next-generation counseling and cancer risk management. Front Oncol 2015;5:1–13.
Li FP, Fraumeni JFJr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969;71:747–752.
Malkin D, Li FP, Strong LC et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1237.
Sorrell A, Espenschied C, Culver J, Weitzel J. Tumor protein p53 (TP53) testing and Li-Fraumeni syndrome: current status of clinical applications and future directions. Mol Diagn Ther 2013;17:31–47.
Masciari S, Dillon DA, Rath M et al. Breast cancer phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort. Breast Cancer Res Treat 2012;133:1125–1130.
Garber JE, Burke EM, Lavally BL et al. Choroid plexus tumors in the breast cancer-sarcoma syndrome. Cancer 1990;66:2658–2660.
Li FP, Fraumeni JF Jr., Mulvihill JJ et al. A cancer family syndrome in twenty-four kindreds. Cancer Res 1988;48:5358–5362.
Chompret A, Abel A, Stoppa-Lyonnet D et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 2001;38:43–47.
Gonzalez KD, Noltner KA, Buzin CH et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009;27:1250–1256.
Birch JM, Hartley AL, Tricker KJ et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994;54:1298–1304.
Bougeard G, Renaux-Petel M, Flaman J-M et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 2015.
Eeles RA. Germline mutations in the TP53 gene. Cancer Surv 1995;25:101–124.
Tinat J, Bougeard G, Baert-Desurmont S et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–109.
Rana HQ, Gelman R, Thompson J et al, Single gene (SG) vs. multi-gene panel (MGP) testing for TP53 germline mutations in Li Fraumeni syndrome (LFS). 65th Annual Meeting of the American Society of Human Genetics, Baltimore, MD, 6–10 October 2015.
Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease—picking up speed. Nat Rev Genet 2017;18:128–142.
Weitzel J, Blazer KR, Laduca H et al, Somatic TP53 mutations detected in germline testing: the importance of phenotypic correlation in cancer predisposition testing (abstr 85). 65th Annual Meeting of the American Society of Human Genetics, Baltimore, MD, 8 October 2015.
Behjati S, Maschietto M, Williams RD et al. A pathogenic mosaic TP53 mutation in two germ layers detected by next generation sequencing. PLoS One. 2014;9:e96531.
Kovar H, Auinger A, Jug G, Muller T, Pillwein K. p53 mosaicism with an exon 8 germline mutation in the founder of a cancer-prone pedigree. Oncogene. 1992;7:2169–2173.
Vazquez-Osorio I, Duat-Rodriguez A, Garcia-Martinez FJ, Torrelo A, Noguera-Morel L, Hernandez-Martin A. Cutaneous and systemic findings in mosaic neurofibromatosis Type 1. Pediatr Dermatol 2017;34:271–276.
Messiaen L, Vogt J, Bengesser K et al. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum Mutat 2011;32:213–219.
Garcia-Romero MT, Parkin P, Lara-Corrales I. Mosaic Neurofibromatosis Type 1: A Systematic Review. Pediatr Dermatol. 2016;33:9–17.
Salo-Mullen EE, Shia J, Brownell I et al. Mosaic partial deletion of the PTEN gene in a patient with Cowden syndrome. Fam Cancer 2014;13:459–467.
Qin L, Wang J, Tian X et al. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J Mol Diagn 2016.
LaDuca H, Stuenkel AJ, Dolinsky JS et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 2014;16:830–837.
Ballinger ML, Best A, Mai PL et al. Baseline surveillance in Li-Fraumeni syndrome using whole-body magnetic resonance imaging: A meta-analysis. JAMA Oncol 2017.
Villani A, Shore A, Wasserman JD et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016.
Pesaran T, Karam R, Huether R et al. Beyond DNA: an integrated and functional approach for classifying germline variants in breast cancer genes. Int J Breast Cancer 2016;2016:2469523.
Castillo D, Herzog J, Sand S, O’Connor T, Clark C, Weitzel J Well-groomed participants: eyebrow plucks as surrogates for biomarker samples and a viable source of constitutional DNA. Annual Meeting of the American Society of Human Genetics, Vancouver, Canada, 18–22 October 2016.
Chompret A, Brugieres L, Ronsin M et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82:1932–1937.
National Comprehensive Cancer Network. NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Version 1.2018. https://www.nccn.org/professionals/physician_gls/recently_updated.asp.
Jaiswal S, Fontanillas P, Flannick J et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–2498.
Steensma DP, Bejar R, Jaiswal S et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
Norquist BM, Harrell MI, Brady MF et al. Inherited mutations in women with ovarian crcinoma. JAMA Oncol 2016;2:482–490.
Swisher EM, Harrell MI, Norquist BM et al. Somatic mosaic mutations in PPM1D and TP53 in the blood of women with ovarian carcinoma. JAMA Oncol 2016;2:370–372.
Coffee B, Cox HC, Kidd J et al. Detection of somatic variants in peripheral blood lymphocytes using a next generation sequencing multigene pan cancer panel. Cancer Genet 2017;211:5–8.
Genovese G, Kahler AK, Handsaker RE et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–2487.
Alix-Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 2016;6:479–491.
Ruark E, Snape K, Humburg P et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–410.
Acknowledgments
The research reported in this article was supported in part by the National Cancer Institute of the National Institutes of Health, under award P30CA033572. The City of Hope Clinical Cancer Genomics Community Research Network was supported in part by award RC4A153828 (PI: J.N.W.) from the National Cancer Institute and the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
E.C.C. was a full-time salaried employee of Ambry Genetics during part of the time she was working on the project (until January 2016), and remains a minority shareholder in the corporation. H.L., T.P., J.S.D., A.E., C.L.G., V.S., and K.J. are full-time salaried employees of Ambry Genetics. The other authors declare no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Weitzel, J.N., Chao, E.C., Nehoray, B. et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med 20, 809–816 (2018). https://doi.org/10.1038/gim.2017.196
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.196
Keywords
This article is cited by
-
SEOM clinical guideline on heritable TP53-related cancer syndrome (2022)
Clinical and Translational Oncology (2023)
-
Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis
Nature Communications (2023)
-
Assessment of genetic susceptibility to multiple primary cancers through whole-exome sequencing in two large multi-ancestry studies
BMC Medicine (2022)
-
Unique roles of rare variants in the genetics of complex diseases in humans
Journal of Human Genetics (2021)
-
Incidental findings from cancer next generation sequencing panels
npj Genomic Medicine (2021)
