
Abstract
Purpose:
This study demonstrates noninvasive prenatal testing (NIPT) for Duchenne muscular dystrophy (DMD) using a newly developed haplotype-based approach.
Methods:
Eight families at risk for DMD were recruited for this study. Parental haplotypes were constructed using target-region sequencing data from the parents and the probands. Fetal haplotypes were constructed using a hidden Markov model through maternal plasma DNA sequencing. The presence of haplotypes linked to the maternal mutant alleles in males indicated affected fetuses. This method was further validated by comparing the inferred single-nucleotide polymorphism (SNP) genotypes to the direct sequencing results of fetal genomic DNA. Prenatal diagnosis was confirmed with amniocentesis, and those results were interpreted in a blinded fashion.
Results:
The results showed an average accuracy of 99.98% for the total inferred maternal SNPs. With a mean depth of 30× achieved in the 10-Mb target region of each sample, the noninvasive results were consistent with those of the invasive procedure.
Conclusion:
This is the first report of NIPT for DMD and the first application of a haplotype-based approach in NIPT for X-linked diseases. With further improvements in accuracy, this haplotype-based strategy could be feasible for NIPT for DMD and even other X-linked single-gene disorders.
Genet Med 17 11, 889–896.
Similar content being viewed by others

Introduction
Duchenne muscular dystrophy (DMD) is the most common inherited neuromuscular disorder and is characterized by noninflammatory but progressive muscle degeneration. It is an X-linked recessive disease that affects 1 in 3,600 to 6,000 live male births.1,2,3 Approximately 70% of DMD cases are caused by deletions/duplications of one or more DMD exons, and 30% of cases have DMD point mutations or small insertions/deletions.4 Deletions/duplications are the most common type of disease-causing mutation of the DMD gene. The importance of prevention has been greatly emphasized because no curative therapy is currently available. Conventional prenatal diagnosis by directly analyzing fetal DNA samples through chorionic villus sampling or amniocentesis represents the current gold standard, but these invasive procedures carry associated procedural risks and cannot be offered to women who decline invasive methods.5,6
The discovery of cell-free fetal DNA (cff-DNA) in the maternal blood circulation has led to new expectations for noninvasive prenatal tests (NIPTs).7,8 For instance, many groups have performed noninvasive screening of fetal aneuploidies, such as trisomy 21, using maternal plasma.9,10 The fetal sex in pregnancies at risk of X-linked genetic disorders and the Rhesus factor status in RhD-negative women represent additional noninvasive applications that have begun to transition from research to routine clinical service.11,12,13 NIPTs for single-gene disorders remain challenging, however, because most (~90%) of the DNA in the maternal plasma is derived from the mother.14
The development of new technologies has further catalyzed the progress of noninvasive tests. In 2008 Lo et al.15 developed a digital polymerase chain reaction (PCR)–based method, called the relative mutation dosage strategy, that determines whether the concentration of the mutant or wild-type allele is overrepresented in the plasma; this method is available for the noninvasive prediction of X-linked disorders.16 This approach is not suitable for NIPTs for DMD, however, because large deletions/duplications cannot be detected. In addition, mutations scattered throughout the 79 exons in the DMD gene without clusters make this locus-specific test laborious and time-consuming. In 2010 Lo et al.17 reported the presence of the entire fetal genome in the maternal plasma and inferred fetal genotypes from the maternal plasma, although they obtained the parental genotypes by sequencing the fetus using DNA that was obtained by invasive sampling. Studies by Kitzman et al.18 and Fan et al.19 introduced accurate, noninvasive fetal genotype inference methods assisted by maternal haplotype but failed to construct a paternal haplotype because of low cff-DNA concentrations. Our group recently developed a new haplotype-based strategy to recover in one step the fetal genotype and haplotype on both parentally heterozygous sites in the maternal plasma.20 Using this approach, we successfully predicted the fetal mutation status of many autosomal recessive disorders based on the maternal plasma, including congenital adrenal hyperplasia, maple syrup urine disease, and congenital deafness.21,22,23 However, the application of this approach to noninvasively predict the fetal mutation status in X-linked diseases has never been demonstrated.
In this study we used our newly developed haplotype-based strategy for DMD mutation detection using target-region sequencing of the maternal plasma. This was the first use of DMD as a representative model system to demonstrate the feasibility of haplotype-based, noninvasive prenatal detection for X-linked disorders.
Materials and Methods
Study design
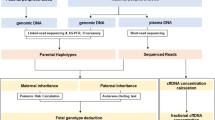
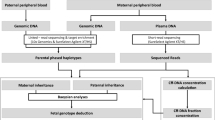
The workflow of this study is illustrated in Figure 1 . NIPTs were blindly performed in parallel with the invasive procedure by two independent groups. Maternal plasma that was collected from at-risk pregnant women was used to noninvasively predict fetal genetic conditions of DMD by target sequencing. An invasive prenatal diagnosis was made based on mutation analysis of fetal genomic DNA that was obtained from amniotic fluid cells. The results of this NIPT method were confirmed by a diagnosis based on prenatal amniocentesis.

The study designation and workflow. First, we collected samples from core families with certain mutations that were identified in the proband and his mother. Then, noninvasive tests and amniocentesis prenatal diagnosis were blindly conducted by two independent groups. Finally, the accuracy of this haplotype-based noninvasive prenatal testing method was evaluated by amniocentesis prenatal diagnosis.
Sample collection and DNA extraction
Eight at-risk families with probands (male patients) were recruited in this study. The causative mutations in the probands and the carrier mothers were previously identified by multiplex ligation-dependent probe amplification (Supplementary Methods online). Informed consent was obtained from all of the participants, and the study protocol was approved by the Ethics Committee of Shanghai Xinhua Hospital. Blood samples (5 ml) were collected from each of the family members (including parents and probands). Maternal peripheral blood samples (10 ml) were collected into two 5-ml ethylenediaminetetraacetic acid–containing tubes before invasive amniocentesis sampling. Maternal plasma was separated from the tubes by initial centrifugation at 1,600×g for 10 min, followed by a second centrifugation at 16,000×g for 10 min. The blood and plasma samples were immediately stored at −80 °C until further processing. Cell-free DNA was isolated from 600 μl of plasma using the TIANamp Micro DNA Kit (Tiangen, Beijing, China). Genomic DNA was extracted from peripheral blood leukocytes using the TIANamp Macro DNA Kit (Tiangen). Fetal DNA was extracted from amniotic fluid cells using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany).
Prenatal diagnosis through amniocentesis sampling
For each family, genomic DNA of the parents, proband, and fetus was used for prenatal analysis. Sanger sequencing and quantitative real-time PCR were used to detect the germ-line mutations and copy-number variants, respectively. The fetal sex was estimated by amplifying the SRY gene and was further confirmed by karyotype analysis. Microsatellites that were closely linked to the DMD gene were used for linkage analysis to determine whether the fetus inherited the maternal mutant allele or the wild-type allele and to exclude maternal contamination.
Design of capture array
A custom-designed 8.81M NimbleGen EZ array containing 2.57M and 6.23M regions in chromosome 22 and chromosome X, respectively, was used for target sequencing on genomic and maternal plasma DNA (Roche NimbleGen, Madison, WI). The probes were randomly distributed on the unique regions in chromosome X, which covered approximately 1.66M exonic regions and 39,319 highly heterozygous single-nucleotide polymorphisms (SNPs), including 1,243 SNPs that were located in the DMD gene region. The other probes covered 0.95M exonic regions on chromosome 22 and 14,345 highly heterozygous SNPs.
Library preparation and massively parallel sequencing
The genomic DNA (gDNA) samples were sonicated (Covaris S2) into fragments of mainly ~200 bp. The fragments were blunted at the ends, and the adaptor was ligated to each end after “A” tailing. Then, the 8-bp barcode was added to each sample through four cycles of PCR for the subsequent massively parallel sequencing. For maternal plasma DNA, the libraries were constructed directly without sonication because of its highly fragmented nature. Other steps similar to those for gDNA were performed, but the number of PCR cycles was increased to 17. A total of 1 µg of the gDNA libraries and 1 µg of the maternal plasma DNA libraries were used for target-region capture following the manufacturer’s instructions. The postcapture libraries were sequenced for 90-bp paired-end reads on a HiSeq 2000 sequencing platform after size distribution, as detected with a DNA bio-analyzer 2100 (Agilent, Santa Clara, CA). The DNA libraries were quantified by quantitative real-time PCR. To achieve sufficient sequencing depth (~28×) in the plasma sequencing, we sequenced seven libraries for each plasma sample.
Sequence alignment and variant calling
The paired-end sequencing reads were mapped to the human reference genome (Hg19, GRCh37) using SOAP2 (ref. 24). PCR duplication or multiple aligned reads were removed. In gDNA sequencing the average duplication rate is ~1.45% for each sample. In plasma sequencing the average duplication rate is ~65.97% for each library. SNP calling was performed using SOAPsnp in the target region25 with two criteria: (i) a depth greater than 8 and (ii) a quality value greater than 20.
Estimation of the fetal gender
The unspecific capture sequences were used to estimate the fetal gender. We predicted the existence of chromosome Y by comparing the ratio of unique map reads between chromosome Y and other autosomes. The threshold was set to 0.5‰ according to the contribution of the candidate cff-DNA concentration and the length ratio between chromosome Y and the autosomes. A ratio less than 0.5‰ indicated a female fetus.
Measurement of the cff-DNA concentration
In the loci on chromosome 22, which were homozygous in both parents but had different types, the fetal genotype was an obligate heterozygote based on Mendel’s laws. In such loci the fetal DNA concentration in the maternal plasma (f) could be calculated using the following equation:  , where p is the count of the fetal inherited, paternal-specific allele and q is the count of the allele that is shared by the fetal and maternal genomes.
, where p is the count of the fetal inherited, paternal-specific allele and q is the count of the allele that is shared by the fetal and maternal genomes.
Haplotype-based NIPT of the fetus at risk of DMD
Because the paternal haplotype and the male proband’s haplotype on chromosome X were readily known from the gDNA genotyping, the key process was the construction of the maternal haplotype on chromosome X, which could be deduced from genotyping of the maternal gDNA and from the proband’s haplotype based on Mendel’s law.20
To infer the fetal haplotype, we used a haplotype-based approach with a linkage relationship that was obtained from the parental haplotypes and allele frequencies that were calculated from the plasma sequencing data, as previously described.20,21,22,23 Because the candidate mutant allele Xa was inherited from the maternal chromosome X, we focused on the fetal maternal-inherited chromosome X in the following analysis process. The informative SNPs that were used to infer fetal haplotype and construct the model were those maternal heterozygous SNP sites on chromosome X. As shown in Figure 2 , a male fetus randomly inherited one of the maternal X chromosome alleles (Xa or Xu), and a female fetus carried a random combination of paternal haplotypes Xp and maternal haplotypes X (Xa or Xu). The fetal genome could then be determined by calculating which of the two possible combinations of the maternal haplotypes were overrepresented in the maternal plasma for male and female fetuses, respectively. According to the recombination characteristics in the process of gametogenesis, we calculated the transmitted probabilities using the distance between the neighbor sites and the sequenced base depth in the plasma. Then, a hidden Markov model (HMM) was established with the parental haplotypes and maternal plasma sequencing data, which were used to calculate the cumulative probability of each of the two possible haplotype combinations. In the process of decoding this HMM chain, we used the Viterbi algorithm to find the most likely sequence of hidden states.20,21,22,23,26 Finally, we could predict the inherited haplotype and recombination breakpoints in the fetus. The detailed calculations that were used to construct the fetal genome and to determine the recombination breakpoints are available in the Supplementary Methods online.

Haplotype-assisted noninvasive prenatal testing (NIPT) method. (a) Target region for enrichment and haplotyping. (b) The haplotype-assisted approach to NIPT for Duchenne muscular dystrophy (DMD). The fetal genome can be one of four combinations that result from the random assortment of parental haplotypes. If a male fetus inherits haplotype Xa (the same as the proband’s, which is the mutant allele) from the mother, this haplotype will be overrepresented in the plasma. By contrast, when haplotype Xu (wild-type allele) is inherited, it exists in greater proportion to haplotype Xa. If a female fetus inherits the maternal Xa haplotype, it would have excessive reads compared with haplotype Xu; by contrast, similar amounts of the two types of maternal haplotypes would exist. Because the father had only one X chromosome, the fetal inherited paternal haplotype can be clearly identified from paternal genotyping. The informative single-nucleotide polymorphisms that were used for analysis were maternal heterozygous sites (*). Based on the maternal haplotypes and maternal plasma sequencing data, the cumulative probability of each of the two possible haplotype combinations in males or females could be calculated using a hidden Markov model. The fetal genetic conditions were predicted by observing the inherited maternal mutant allele, which was then used for NIPTs for DMD.
Results
Family cases
For families F01–F08, the causative mutations were identified as deletions of exons 20–23, deletions of exons 45–48, a duplication of exon 2, c.9760_9781dupTTTGGGGGCAGTAACATTGAGC, deletions of exons 49–52, a duplication of exon 56, a deletion of exon 17, and a deletion of exon 45, respectively (Supplementary Figure S1 online). All pregnant mothers were DMD carriers with a gestational age ranging from 17–22 weeks. The pedigrees and the DMD mutations identified in the eight families are shown in Supplementary Figure S1 online.
Prenatal diagnosis through amniotic sampling
For families F02 and F08, the fetuses were females who did not inherit the maternal mutations. For family F07, the fetus was a female that carried the same mutation as that identified in the proband. The fetuses in the remaining five families were all male patients. Linkage studies based on short tandem repeat genotyping were compatible with the molecular diagnosis. Maternal contamination was excluded based on the microsatellite information (data not shown).
Measurements of the fetal gender, cff-DNA concentration, and sequencing error rate
The data that were used for the following analysis are shown in Table 1 . In these samples, the relative unique map ratio of chromosome Y in male fetuses was much higher than that in female fetuses. For families F02, F07, and F08, the ratio of unique map reads between chromosome Y and the autosomes was 0.44, 0.45, and 0.39‰, respectively, indicating a female fetus in the current pregnancy. For families F01 and F03–F06, the ratio varied from 0.68 to 2.42‰, indicating the existence of chromosome Y; we thus predicted these fetuses as being male.
The cff-DNA concentrations in the maternal plasma were calculated using the SNP sites that were homozygous in both parents but had different types on chromosome 22. For these eight families, the cff-DNA concentrations ranged from 3.52 to 22.67%, showing significant differences between individuals.
We calculated the sequencing error rate using the SNPs that were homozygous with the same genotype in both parental genomes on chromosome 22 but had different bases in the plasma. The overall sequencing error rate was ~0.14% (Supplementary Materials online).
Construction of parental haplotype on chromosome X
The overall methodology for the NIPT of DMD is shown in Figure 1 . Following the NIPT flowchart, the maternal haplotypes could be constructed based on the genotyping information from the probands, and the paternal haplotypes could be directly obtained from the genotyping by genomic sequencing. Clean data with more than 20-fold coverage were obtained for each gDNA sample, with an average of 95.23% coverage and a mean depth of 36.40 in the target regions. A mean of 7,234,531 SNPs on chromosome X were detected and constructed using a trio-based strategy in the gDNA samples from the parents and the proband; these were used in the HMM to predict fetal haplotypes ( Table 2 ; Supplementary Table S2 online).
NIPT using maternal plasma sequencing
We obtained a total of 97.57 million raw reads for each sample of maternal plasma DNA, with a mean depth of 28.05 and coverage of 95.91% in the targeted region ( Table 2 ). On the basis of the parental haplotype data and the maternal plasma DNA sequencing data, the fetal haplotype was inferred using the HMM. The number of informative SNPs phased on chromosome X for fetal haplotype construction ranged from 2,590 to 7,556 for each family. The HMM-based prediction of recombination and the predicted fetal haplotype are shown in Figure 3a (using family F07 as an example). In the genomic region around the DMD gene, the fetus inherited the maternal haplotype Xa linked to the mutant allele. Given that the fetal gender was predicted to be female, the fetus was thus predicted to be a carrier of the DMD mutation.

Noninvasive prenatal testing (NIPT) using maternal plasma DNA by massively parallel sequencing. (a) The hidden Markov model–based prediction of recombination and the fetal inherited haplotype in family F07. The x-axis represents loci on chromosome X, the right y-axis represents the logarithmic values of the odds ratios of fetal inheritance of the maternal haplotypes, and the left y-axis represents the value of 1 minus the allele frequency of haplotype Xa (1 − Xa). The orange line represents the maternal allele. (b) NIPT results for eight families. The x-axis and y-axis indicate the target region surrounding the DMD gene on chromosome X and the odds ratio of the fetal inherited haplotype in the maternal plasma, respectively. The fetal inherited haplotypes for eight families are depicted in lines of different colors. An odds ratio less than zero indicates that the fetus inherited the maternal wild-type haplotype, whereas a value of more than zero indicates inheritance of the mutant haplotype.
All NIPT results for eight families are shown in Figure 3b . The results were corrected by the odds ratio of the two types of maternal inherited haplotypes; an odds ratio less than zero indicated that the fetus inherited the same haplotype as the proband (Xa), which was linked to a maternal mutant-type allele, whereas a value greater than zero indicated that the fetus inherited the opposite allele (Xu). Thus we predict that F02 and F08 inherited the maternal wild-type haplotypes, whereas the others inherited the mutant allele from Figure 3b . Considering the gender information, F02 and F08 should not be susceptible to DMD, F07 should be an unaffected carrier, and the others would be male fetuses affected with DMD. These results are consistent with the prenatal diagnosis obtained through amniocentesis.
Accuracy of the inferred fetal haplotype
To estimate further the accuracy of the inferred fetal haplotype for NIPT, the SNP genotypes that were inferred through maternal plasma DNA sequencing were validated using direct sequencing of fetal gDNA that was collected from amniotic fluid cells. Combining the genomic sequencing data of the parents and the fetuses, the standard fetal haplotypes could be constructed based on the trio strategy, as previously described.20,21,22,23 Because the father has one X chromosome, we compared the accuracy of only the fetal inferred maternal SNP sties with the standard data obtained by direct sequencing. The total phased maternal SNP sites on chromosome X for each family showed a concordant ratio >99.98% (Supplementary Table S3 online). The accuracy of the inferred heterozygous SNP sites was relatively low, which may be caused by the low sequencing depth and the recombinant events that occurred on chromosome X. The issue of balancing the sequencing depth and the accuracy of the inferred heterozygous SNPs remains a problem and should be addressed in future studies.
Discussion
New technologies (such as digital PCR and next-generation sequencing) have greatly accelerated the development of NIPT in clinical applications, including fetal gender estimation, RhD status determination, and aneuploidy screening.9,10,11,12,13 Most studies thus far, however, have been restricted to the identification of paternal-specific sequences.27 The ability to predict noninvasively whether the genetic variants that are passed from the mother to the fetus are at risk of single-gene disorders remains challenging, mainly because of the high background of maternal DNA in the plasma mixture that prevents an accurate prediction of fetal status. Few studies of the noninvasive diagnosis of single-gene disorders have been reported to date, especially those investigating X-linked diseases.
Our recently developed haplotype-assisted strategy provides a new opportunity for NIPT for monogenic diseases.20 Combining the deep sequencing of samples from the parent, proband, and maternal plasma, the fetal haplotype could be deduced using an HMM model with the assistance of the parental haplotypes (deduced from the proband’s sample). The fetal mutation status was estimated according to whether the haplotype that was linked to the mutant allele or the wild-type allele was inherited, which is the principle of NIPT for single-gene disorders based on a haplotype-assisted strategy. Using this approach, we noninvasively diagnosed fetuses at risk of congenital adrenal hyperplasia, maple syrup urine disease, and congenital deafness with high accuracy.21,22,23 However, these disorders are inherited in an autosomal recessive pattern, and our current report is the first to apply a haplotype-assisted strategy to NIPT for X-linked disorders. In addition, most studies have focused on demonstrating the feasibility of NIPT for single-gene disorders in case reports, whereas this study is the first to perform NIPT in parallel with invasive prenatal diagnosis in a small pilot sample, and our results have important significance for clinical practice.
For NIPT for X-linked disorders, the availability of the digital relative mutation dosage strategy and our haplotype-based approach allows researchers to choose the appropriate strategy for their applications in different fields. As discussed in the article by Lam et al.,28 the former strategy is more simple and cost-effective for targeting one or a limited number of loci. In addition, its classification of at-risk fetus depends on the information of the cff-DNA concentration, the absence of unique fetal-specific markers results in failure of the measurement of cff-DNA concentration for female fetuses. For example, although Barrett et al.29 correctly classified 75% of female fetuses (15 of 20) as at risk for the sickle cell genotype, they stated that these markers were informative for only 65% of the female-bearing pregnancies. By contrast, a haplotype-based strategy permits NIPT in both male and female pregnancies using linkage relationships. While the cost of deep sequencing is still an issue for wide application, the balance between efficiency and cost needs to be addressed.
Although the noninvasive prenatal diagnosis of DMD using a haplotype-based approach provided results consistent with those of the amniotic prenatal diagnosis, there remain several avenues for improvement. The first limitation is the impact of the fetal concentration on the accuracy of the results. In fact, the use of a site-by-site strategy according to the observation of allelic imbalance at individual sites is ideal and easy for the inference of paternal-specific transition. However, this strategy is largely dependent on the concentration of cff-DNA and the sequence depth. Kitzman et al.18 also demonstrated this point: In the trio G1 (whole-genome sequencing ~56× at 8.14 weeks) with a low cff-DNA concentration (Supplementary Materials online), only 60.3% of the paternal-specific alleles were identified. Other studies showed that the minimum fetal cff-DNA required for analysis to report a reliable clinical result was ~4%.30 In this study, at concentrations of 3.52 and 4.81%, we were able to accurately predict the inheritance of the maternal allele using a haplotype-based approach without typically increasing the sequencing depth. Nevertheless, additional subjects should be used to evaluate the performance of this strategy at a low fetal fraction to support noninvasive tests in the early gestational weeks when the concentration of cff-DNA is very low.31
The second avenue for improvement is the accuracy of predicting the location of the recombination disruption. Because recombination events can affect the prediction accuracy of NIPT based on haplotype analysis, especially for cases where the mutation could be located in relation to a recombination breakpoint, researchers should pay much attention to the fetal haplotype construction and recombination breakpoint determination for these cases. Lo et al.17 demonstrated that haplotype-based analysis could potentially detect maternal recombination, but the observed change was located before the actual artificial recombinant site. In this study, partial inheritance in a small minority of blocks (0.6%; n = 72) corresponding to switch errors from the haplotype assembly and to sites of recombination was observed in family F08, which was predicted to occur at chrX 33,040,143–33,244,129, ~1.16 Mb away from the germ-line mutation that occurred in exons 45–49 of the DMD gene. Thus we predicted that the fetus inherited the maternal “not-at-risk” haplotype in a disease-causing region, ruling out the possibility of inheriting the deletion mutation that was carried by her mother. Invasive prenatal testing also confirmed this result. The recombination frequency expected for the dystrophin gene based on its size (2.4 Mb) is 2.4%; however, the observed frequency was higher (6–10%) than expected according to previous reports.32,33,34,35 How to accurately infer the fetal inherited allele for recombination cases remains an area for future effort. With the development of new bioinformatics models and improvements in haplotyping, we believe that noninvasive prenatal testing could provide a preliminary screening approach for most at-risk pregnant women. In clinical practice genetic counseling should fully consider the error risk caused by recombination, and invasive prenatal diagnosis through chorionic villus sampling or amniocentesis sampling is still suggested to provide recombination cases with a higher degree of certainty.
The third avenue for improvement is the estimation of fetal gender. Here we correctly predicted the existence of chromosome Y for eight fetuses from unspecific sequences, and many other approaches could also be used to noninvasively determine the fetal gender based on maternal plasma. Indeed, several reports of noninvasive fetal sex determination using PCR-based amplification of Y-chromosome-specific sequences, such as the DYS marker or the SRY gene,11,12 have been published, and this method is reliable, with high sensitivity and specificity, as confirmed by numerous studies.36,37 Considering the factors of cost and time, our future work will aim to develop specific Y chromosome probes on the capture array or to detect specific sequences on chromosome Y that can be used to estimate the fetal gender.
Finally, the turnaround time and cost of massively parallel sequencing–based NIPTs remain challenging issues that need to be addressed. The universal platform of HiSeq 2000 requires a run time of ~11 days. With the arrival of the HiSeq 2500, however, a rapid run can be conducted within 40 h, making it possible to produce results even faster.38 With further developments in high-throughput sequencing technologies, we believe that the cost of such techniques will greatly decrease in the coming years.
This first application of the haplotype-based strategy to DMD prenatal detection expanded the coverage of NIPT for monogenic disorders to X-linked diseases and will undoubtedly have profound implications for the future of prenatal genetic diagnostics. Our NIPT for DMD provided information regarding fetal sex, which could serve as a first-tier screening test to significantly reduce unnecessary invasive procedures in female pregnancies. Second, the NIPT method is a simple procedure that may be easier to access than invasive testing, as it is immediately accessible by blood test. Finally, NIPTs permit earlier testing; if an effective haplotype-based strategy at a low fetal fraction was available after further large-sample testing, NIPT for DMD would permit testing during the early weeks of gestation, increasing the time required to make a decision about terminating the pregnancy. In addition, it can provide an option for women who decline invasive methods to get better information about the likelihood of an affected fetus. Although there are many advantages compared with invasive methods, the accuracy of NIPT for DMD (especially in recombination cases) still needs to be improved before use in clinical situations. With improved accuracy and lower cost, we speculate that NIPT will serve as an alternative for faster and easier prenatal detection of pregnant women at risk of having fetuses with DMD and other X-linked disorders in the future.
Disclosure
The authors declare no conflict of interest.
References
Drousiotou A, Ioannou P, Georgiou T, et al. Neonatal screening for Duchenne muscular dystrophy: a novel semiquantitative application of the bioluminescence test for creatine kinase in a pilot national program in Cyprus. Genet Test 1998;2:55–60.
Bradley D, Parsons E . Newborn screening for Duchenne muscular dystrophy. Semin Neonatol 1998;3:27–34.
Emery AE . Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord 1991;1:19–29.
Takeshima Y, Yagi M, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 2010;55:379–388.
Evans MI, Krivchenia EL, Wapner RJ, et al. Principles of screening. In: Evans MI (ed). Clinical Obstetrics and Gynecology: New Genetics for the Clinician. Lippincott, Williams & Wilkins: Philadelphia, PA, 2002:657–660.
Mujezinovic F, Alfirevic Z . Procedure-related complications of amniocentesis and chorionic villous sampling: a systematic review. Obstet Gynecol 2007;110:687–694.
Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet 1997;350:485–487.
Lo YM . Fetal DNA in maternal plasma/serum: the first 5 years. Pediatr Res 2003;53:16–17.
Mersy E, Smits LJ, van Winden LA, et al.; South-East Netherlands NIPT Consortium. Noninvasive detection of fetal trisomy 21: systematic review and report of quality and outcomes of diagnostic accuracy studies performed between 1997 and 2012. Hum Reprod Update 2013;19:318–329.
Papageorgiou EA, Patsalis PC . Non-invasive prenatal diagnosis of aneuploidies: new technologies and clinical applications. Genome Med 2012;4:46.
Bustamante-Aragones A, Rodriguez de Alba M, Gonzalez-Gonzalez C, et al. Foetal sex determination in maternal blood from the seventh week of gestation and its role in diagnosing haemophilia in the foetuses of female carriers. Haemophilia 2008;14:593–598.
Devaney SA, Palomaki GE, Scott JA, Bianchi DW . Noninvasive fetal sex determination using cell-free fetal DNA: a systematic review and meta-analysis. JAMA 2011;306:627–636.
Freeman K, Szczepura A, Osipenko L . Non-invasive fetal RHD genotyping tests: a systematic review of the quality of reporting of diagnostic accuracy in published studies. Eur J Obstet Gynecol Reprod Biol 2009;142:91–98.
Lun FM, Chiu RW, Chan KC, Leung TY, Lau TK, Lo YM . Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem 2008;54:1664–1672.
Lun FM, Tsui NB, Chan KC, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci USA 2008;105:19920–19925.
Tsui NB, Kadir RA, Chan KC, et al. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood 2011;117:3684–3691.
Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91.
Kitzman JO, Snyder MW, Ventura M, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012;4:137ra76.
Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR . Non-invasive prenatal measurement of the fetal genome. Nature 2012;487:320–324.
Chen SP, Ge HJ, Wang XB, et al. Haplotype-assisted accurate non-invasive fetal whole genome recovery through maternal plasma sequencing. Genome Med 2013;5:18.
Ma DY, Ge HJ, Li XC, et al. Haplotype-based approach for noninvasive prenatal diagnosis of congenital adrenal hyperplasia by maternal plasma DNA sequencing. Gene 2014;2:252–258.
You Y, Sun Y, Li X, et al. Integration of targeted sequencing and NIPT into clinical practice in a Chinese family with maple syrup urine disease. Genet Med 2014;16:594–600.
Meng M, Li XC, Chen F, et al. Noninvasive prenatal testing for autosomal recessive conditions by maternal plasma sequencing in a case of congenital deafness. Genet Med 2014;16:972–976.
Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009;25:1966–1967.
Li R, Li Y, Fang X, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 2009;19:1124–1132.
Tanaka H, Ishikawa M, Asai K, Konagaya A . Hidden Markov models and iterative aligners: study of their equivalence and possibilities. Proc Int Conf Intell Syst Mol Biol 1993;1:395–401.
Wright CF, Burton H . The use of cell-free fetal nucleic acids in maternal blood for non-invasive prenatal diagnosis. Hum Reprod Update 2009;15:139–151.
Lam KW, Jiang P, Liao GJ, et al. Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to β-thalassemia. Clin Chem 2012;58:1467–1475.
Barrett AN, McDonnell TC, Chan KC, Chitty LS . Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem 2012;58:1026–1032.
Wang E, Batey A, Struble C, Musci T, Song K, Oliphant A . Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat Diagn 2013;33:662–666.
Lo YM, Tein MS, Lau TK, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 1998;62:768–775.
Oudet C, Hanauer A, Clemens P, Caskey T, Mandel JL . Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum Mol Genet 1992;1:599–603.
Nobile C, Galvagni F, Marchi J, Roberts R, Vitiello L . Genomic organization of the human dystrophin gene across the major deletion hot spot and the 3’ region. Genomics 1995;28:97–100.
Ferreiro V, Giliberto F, Francipane L, Szijan I . The role of polymorphic short tandem (CA)n repeat loci segregation analysis in the detection of Duchenne muscular dystrophy carriers and prenatal diagnosis. Mol Diagn 2005;9:67–80.
Giliberto F, Ferreiro V, Massot F, Ferrer M, Francipane L, Szijan I . Prenatal diagnosis of Duchenne/Becker muscular dystrophy by short tandem repeat segregation analysis in Argentine families. Muscle Nerve 2011;43:510–517.
Fernández-Martínez FJ, Galindo A, Garcia-Burguillo A, et al. Noninvasive fetal sex determination in maternal plasma: a prospective feasibility study. Genet Med 2012;14:101–106.
Wright CF, Wei Y, Higgins JP, Sagoo GS . Non-invasive prenatal diagnostic test accuracy for fetal sex using cell-free DNA a review and meta-analysis. BMC Res Notes 2012;5:476.
Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012;2012:251364.
Acknowledgements
This work was funded by the Foundation from the Institute of Shanghai Municipal Health (20134Y037 and 20134094). We thank the subjects and their families for participating. We also acknowledge the coinvestigators for their help in the study design, data analysis, and sample acquisition, and Y. Sun for revising the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
(DOC 1374 kb)
Rights and permissions
About this article

Cite this article
Xu, Y., Li, X., Ge, Hj. et al. Haplotype-based approach for noninvasive prenatal tests of Duchenne muscular dystrophy using cell-free fetal DNA in maternal plasma. Genet Med 17, 889–896 (2015). https://doi.org/10.1038/gim.2014.207
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.207
Keywords
This article is cited by
-
Clinical evaluation of non-invasive prenatal screening for the detection of fetal genome-wide copy number variants
Orphanet Journal of Rare Diseases (2022)
-
Noninvasive prenatal diagnosis of duchenne muscular dystrophy in five Chinese families based on relative mutation dosage approach
BMC Medical Genomics (2021)
-
Noninvasive prenatal diagnosis of monogenic disorders based on direct haplotype phasing through targeted linked-read sequencing
BMC Medical Genomics (2021)
-
Noninvasive prenatal testing for β-thalassemia by targeted nanopore sequencing combined with relative haplotype dosage (RHDO): a feasibility study
Scientific Reports (2021)
-
The evolving role of genetic tests in reproductive medicine
Journal of Translational Medicine (2019)
