
Abstract
Purpose:
Spinal muscular atrophy is a common autosomal-recessive disorder caused by mutations of the SMN1 gene. Spinal muscular atrophy carrier screening uses dosage-sensitive methods that determine SMN1 copy number, and the frequency of carriers varies by ethnicity, with detection rates ranging from 71 to 94% due to the inability to identify silent (2 + 0) carriers with two copies of SMN1 on one chromosome 5 and deletion on the other. We hypothesized that identification of deletion and/or duplication founder alleles might provide an approach to identify silent carriers in various ethnic groups.
Methods:
SMN1 founder alleles were investigated in the Ashkenazi Jewish population by microsatellite analysis and next-generation sequencing.
Results:
An extended haplotype block, specific to Ashkenazi Jewish SMN1 duplications, was identified by microsatellite analysis, and next-generation sequencing of SMN1 further defined a more localized haplotype. Of note, six novel SMN1 sequence variants were identified that were specific to duplications and not present on single-copy alleles. The haplotype was also identified on SMN1 duplication alleles in additional ethnic groups.
Conclusion:
Identification of these novel variants in an individual with two copies of SMN1 significantly improves the accuracy of residual risk estimates and has important implications for spinal muscular atrophy carrier screening.
Genet Med 16 2, 149–156.
Similar content being viewed by others


Introduction
Spinal muscular atrophy (SMA) is one of the most common autosomal-recessive diseases, with an incidence of ~1 in 10,000 live births and a carrier frequency of 1 in 35–117, depending on ethnicity.1 The disease is characterized by the progressive degeneration and loss of anterior horn cells in the spinal cord and brainstem nuclei causing symmetric muscle weakness and atrophy,2,3 and the clinical subtypes (I–IV) are primarily based on age at onset and disease severity.
The major cause of SMA is homozygous deletion of the SMN1 gene on chromosome 5q13.2.4 In 95–98% of SMA patients, both copies of SMN1 exons 7 are either deleted or are rendered nonfunctional due to gene conversion.5,6 The remaining 2–5% of patients carry intragenic mutations and are compound heterozygotes.7 SMN1 and SMN2 are separated by ~1.4 Mb of sequence within a segmental duplication on 5q13.2. They have high sequence similarity,8,9 and there are no encoded amino acid differences. A single base change affecting a putative splice enhancer in exon 7 (c.840C>T) accounts for splicing differences such that the majority of SMN2 tran scripts lack exon 7.10,11 Some genotype/phenotype correlations have been established among patients who carry SMN1 point mutations, and in general, the presence of additional copies of SMN2 positively modifies clinical prognosis.12,13
The majority of mutations causing all SMA subtypes involve SMN1 copy-number loss. Consequently, carrier screening must be performed by dosage-sensitive methods that can distinguish SMN1 from SMN2, including quantitative PCR,14 multiplex ligation-dependent probe amplification (MLPA),15 and/or TaqMan quantitative technology.16 Of note, none of these established methods can determine the number of SMN1 copies present on individual chromosomes. Individuals with two SMN1 copies on one chromosome (duplication allele) and no copies on the other (deletion allele) are silent (2 + 0) carriers as most individuals with two intact SMN1 copies (1 + 1) are not carriers ( Figure 1 ).17 Therefore, SMA carrier detection by current techniques generates false-negative results.18 The frequency of silent (2 + 0) carriers varies and is directly proportional to the product of the deletion and duplication allele frequencies in a given population.

Schematic of SMN1 and SMN2 alleles on chromosome 5q13. (a) Wild type with one copy of SMN1 and SMN2 on each chromosome 5 (1 + 1). (b) SMA carrier with one copy of SMN1 on one chromosome 5 and loss of SMN1 on the other chromosome (1 + 0). (c) Duplication with two copies of SMN1 on one chromosome 5 and one copy on the other chromosome (2 + 1). (d) SMA silent carrier with two copies of SMN1 on one chromosome 5 and loss of SMN1 on the other chromosome (2 + 0).
Among Ashkenazi Jewish (AJ) individuals, the SMA carrier frequency has been reported as 1 in 41, with a detectability of ~90%.1 Of the remaining 10%, ~8% are silent (2 + 0) carriers, and the rest are compound heterozygotes with deletion of one allele and intragenic mutation in the other.7 In addition, ~2% of SMA patients have a de novo SMN1 mutation with only one carrier parent.6 The frequency of SMN1 silent (2 + 0) carriers in any population modifies the residual risk of being a carrier after a negative screening result. The most striking example can be found in the African-American (AA) population, where the frequency of duplication (2 + 1) individuals is relatively high (47%), resulting in a greater number of silent (2 + 0) carriers, such that the carrier detection rate is only 71%, with a residual risk of 1 in 121 after a negative dosage screening result.1
Because the ability to identify silent (2 + 0) carriers would significantly improve carrier detection, efforts were directed to identify ethnic-specific SMN1 founder deletion and/or duplication alleles by detecting a genotype unique to either the deletion or duplication alleles present in silent (2 + 0) carriers. The feasibility of this approach for the AJ population has been supported by the fact that carrier screening in this population is based on the occurrence of common recessive disease mutations due to shared haplotype blocks with founder mutations.19,20
We identified AJ founder alleles for SMN1, including one that is present in approximately half of all AJ SMN1 duplications (2 + 1). Sequencing across the SMN1 genomic region identified six novel variants and confirmed that a unique haplotype was common to both AJ and AA duplication alleles. Identification of these variants can improve carrier detection and provide more accurate estimates of residual risk with respect to SMA carrier status.
Materials and Methods
Specimen collection and DNA extraction
Genomic DNA was obtained with informed consent from the peripheral blood specimens of anonymous individuals living in the Greater New York Metropolitan area, who self-identified as AJ, AA, Hispanic, or Caucasian (CA). DNA was isolated using the Puregene DNA Purification kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
MLPA
A total of 200 ng of genomic DNA from each specimen was used for the MLPA analysis with the Salsa MLPA SMA P021 kit (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer’s instructions.21 The MLPA PCR products were separated by capillary electrophoresis using the ABI-3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA). The results were imported into GeneMarker software (Softgenetics, State College, CA) for the MLPA data analysis using population normalization with the MLPA ratio as the analysis method and peak height as the quantification method.
Microsatellite analysis
Microsatellite analysis was performed with markers flanking the SMN genes on 5q13, which included D5S681, D5S435, and D5S61022 and three novel markers, MS1, MS2, and MS3, created based on publicly available human genomic sequence (http://genome/ucsc.edu). Each primer pair was amplified separately in a total volume of 25 µl with genomic DNA (see Supplementary Tables S1–S3 online). The PCR products were then run on the ABI-3130XL Genetic Analyzer, and the results were analyzed with GeneMapper software (Applied Biosystems).
Long-range PCR and sequencing
The region between exons 7 and 8 of the SMN1 gene in AJ duplication carriers was amplified using Long-Range PCR reagents (Qiagen) with primers SMN1-E7F and SMN1-E8R. Primer SMN1-E7F was designed to preferentially amplify SMN1 by the inclusion of c.840C at exon 7. PCR amplification was performed according to the manufacturer’s instructions. The PCR amplicons were then sequenced using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) on an ABI 3730 DNA analyzer using the same PCR primers and two additional primers, SMN1-I7F2 and SMN-E8Ra, to confirm sequencing results in both directions (see Supplementary Tables S1–S3 online).
TA cloning
LongRange PCR fragments from five AJ major duplication (2 + 1) carriers were subjected to TA cloning. In brief, amplicons were gel purified (Qiagen) and cloned into the PCR vector from the TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Twelve clones from each of these individuals were isolated for PCR screening. Clones with the expected insert size were subsequently subcultured, and plasmids were purified using the QIAprep Spin Miniprep Kit (Qiagen) and sequenced as described above using M13 primers (Fisher Scientific, Pittsburgh, PA).
Restriction fragment length polymorphism analysis
PCR was performed to amplify the fragment containing g.27134T>G alteration with the primers SMN1-E7F and SMN1-I7R1 (see Supplementary Tables S1–S3 online). Five microliters of product were digested with 4 U of HypCH4III (New England Biolabs, Ipswich, MA) at 37 °C for 4 h and were resolved by 1.5% agarose gel electrophoresis.
Targeted next-generation sequencing of SMN1
The target SMN1 region was amplified with a set of five primer pairs (see Supplementary Tables S1–S3 online) using a SequalPrep Long PCR Kit (Invitrogen) according to the manufacturer’s instructions. The PCR products were normalized with a SequalPrep Normalization Plate Kit (Invitrogen). Normalized PCR fragments from each amplicon were pooled together and were treated according to the standard Illumina sequencing protocols. DNA libraries were sequenced with Illumina GAIIx (Illumina, San Diego, CA) in one lane for 36 cycles.
Base calls and quality scores were generated using NextGENE software (SoftGenetics). The resulting FASTA files were aligned to the SMN1 reference sequence (NCBI build 37.2). A mutation report with a list of variant calls and corresponding quality metrics was generated. Variations from 12 samples were grouped and compared. Only variations specific to each group were kept and further validated by Sanger sequencing as described above.
Statistical analysis for microsatellite markers
Genotype frequencies for each microsatellite marker were tested for Hardy–Weinberg equilibrium using the χ2 test. Haplotypes of the markers, D5S681-D5S435-MS1-D5S610, were estimated for AJ controls and AJ SMN1 deletions (1 + 0) and duplications (2 + 1) using SAS/Genetics version 9.2 (SAS Institute, Cary, NC). Putative haplotypes were inferred from their frequency among all examined individuals and reconstructed based on the most likely combination of alleles. Differences in haplotype frequencies between the AJ control cohort and the AJ cohort of individuals with SMN1 deletions or duplications were tested using the χ2 test. P values for overall and pairwise comparisons were calculated for the four-marker haplotypes, with the threshold of 0.05 considered statistically significant. Improvement in the detection rate with the addition of sequence variants was estimated using the Bayesian approach (see Supplementary Methods and Procedures online).
Results
AJ population screening for SMN1 deletions and duplications
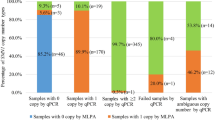
To determine the frequency of SMN1 deletions (1 + 0) and duplications (2 + 1) in the AJ population, 692 individuals of 100% AJ ancestry (with four AJ grandparents) were screened by an SMN1- and SMN2-specific MLPA assay.15,21 Fifteen SMN1 deletion (1 + 0) carriers (1 in 46 or 2.2%) were identified, which is similar to the frequency reported in other European populations.23 Ninety-nine individuals with SMN1 duplications (2 + 1) (14.3%) were identified, but none had four SMN1 copies (i.e., two duplication alleles, 2 + 2), although the expected frequency of (2 + 2) individuals was 0.5% (see Supplementary Table S4 online). The observed frequencies did not deviate significantly from Hardy–Weinberg equilibrium (exact P > 0.05).
Frequency of AJ SMA carriers
Using the frequency of deletions (1 + 0) (2.2%) and duplications (2 + 1) (14.3%), assuming Hardy–Weinberg equilibrium for three alleles (SMN1 = 0, 1, or 2 copies), silent (2 + 0) carriers were estimated to occur at a frequency of 2qr (where q is the frequency of 0 copies and r is the frequency of 2 copies), which was 1 in 531 AJ individuals. On the basis of this calculation, the overall carrier frequency for SMA among AJ individuals was ~1 in 41.1 (2.4%), which includes carriers with loss of one SMN1 (1 + 0) copy, silent (2 + 0) carriers, and the small fraction of carriers with intragenic mutations. The carrier detection rate in the AJ population by MLPA was ~90% on the basis of this analysis, with ~8% silent (2 + 0) carriers and ~2% intragenic mutation carriers. Therefore, the residual risk of being a carrier after a negative MLPA screening result with two copies of SMN1 was 1 in 345 ( Table 1 ).
Mutation groups among AJ individuals with deletions (1 + 0) or duplications (2 + 1)
Twenty-three AJ deletion (1 + 0) carriers were identified and fell into one of four major mutation groups: 1del, SMN1 deletion only; 2del, SMN1 and SMN2 deletions; 3del, SMN1 exons 7 and 8 deletion and SMN2 deletion, or an SMN1 deletion and SMN2 exons 7 and 8 deletion; and 4del, SMN1 exons 7 and 8 deletion only (see Supplementary Table S5 online).
Seventy-two AJ individuals with duplications (2 + 1) were detected by MLPA, and three major groups were identified: 1dup, three SMN1 copies with no SMN2 copy; 2dup, three SMN1 copies with one SMN2 copy, and 3dup, three SMN1 copies with two SMN2 copies (see Supplementary Table S5 online). Of the 72 SMN1 duplications examined, 26 (36.1%) had no SMN2 copies, and an additional 32 (44.4%) had one SMN2 copy, suggesting that the duplication in these 58 individuals arose by a gene conversion event. Another 11 (15.3%) SMN1 duplication individuals had two SMN2 copies, and it is likely that those duplications arose by a different mechanism, such as nonallelic homologous recombination24 or, alternatively, may have resulted from a gene conversion event in individuals with SMN2 duplications.25
Haplotype reconstruction in AJ individuals with deletions (1 + 0) or duplications (2 + 1)
To determine whether founder haplotypes were present for SMN1 deletion alleles, 20 carriers from the groups 1del, 2del, and 3del were genotyped with six microsatellite markers flanking the SMN1 and SMN2 genes, including D5S681, D5S435, and D5S61022 and three novel markers, MS1, MS2, and MS3 (see Supplementary Figure S1 online). Seventy-eight AJ control individuals with two SMN1 copies were also genotyped. Four consecutive loci D5S681-D5S435-MS1-D5S610 were then chosen for marker association studies. Their most likely haplotypes were constructed using SAS/Genetics version 9.2 for the 20 SMN1 deletion carriers including all individuals from groups 1del, 2del, and 3del ( Table 2 ). Group 4del was excluded because all three individuals only had loss of exons 7 and 8.
Two putative founder haplotypes were significantly enriched (P ≤ 0.001) in the deletion carriers as compared with controls. The most common allele combination among the 20 carriers, 2-5-4-5, was assigned in 22.5% of carriers and was possible, although not assigned by the software, in 8 additional (40%) carriers. The 2-5-4-5 haplotype was also assigned to one chromosome of 78 controls (0.6%), indicating that this haplotype may not be specific to deletion alleles. The other carrier-specific allele haplotype, 2-1-4-5, was not assigned to any control individuals; however, two control individuals could potentially carry it. One of the group 4del carriers with the SMN1 exons 7 and 8 deletion only could possibly have the 2-5-4-5 haplotype, indicating that it may not be specific to SMN1 whole-gene copy-loss alleles or that it may be the haplotype of the wild-type allele in carrier no. 23 (see Supplementary Table S5 online).
To analyze the individuals with duplications (2 + 1) for unique shared haplotypes, the six microsatellite markers were genotyped in 69 of the 99 AJ individuals with three copies of SMN1 and in 3 additional AJ individuals who were subsequently identified with four copies of SMN1 (see Supplementary Table S5 online). The same four markers, D5S681-D5S435-MS1-D5S610, were used in haplotype reconstruction for 42 individuals with duplications from groups 1dup and 2dup. Of these, 26 had three SMN1 copies and no SMN2 copies, and 16 had three SMN1 copies and one SMN2 copy. A separate analysis was performed for the nine 3dup group individuals who carried three SMN1 copies and two SMN2 copies, because they presumably represented a separate duplication group.
The most likely marker haplotypes for the three groups are shown in Table 2 . One major duplication marker haplotype, 2-5-6-10, was significantly enriched in groups 1dup and 2dup (19%). This haplotype was completely absent from the controls and was highly specific for the duplication carriers (P < 0.0001). Of the 58 1dup and 2dup group individuals, 23 (39.7%) had the full four-marker haplotype and eight (13.8%) had the 2-5-6 partial haplotype. In addition, one of the 3dup group individuals (no. 59; see Supplementary Table S5 online) had the 2-5-6-10 haplotype, indicating that this individual most likely carried the SMN2 duplication on the wild-type allele. The major duplication haplotype was also positive in two of three individuals with four SMN1 copies, of which one individual was homozygous for the 5-6-10 haplotype (see Supplementary Table S5 online).
Sequencing of the major AJ duplication allele
Several putative AJ founder haplotypes were computationally inferred in the SMN1 deletion and duplication alleles. The major duplication haplotype, 2-5-6-10, or the partial haplotype, 2-5-6, was present in the majority (51.7%) of AJ duplications with 0 or 1 copy of SMN2 (see Supplementary Table S5 online) and was absent from the AJ control population. Therefore, this duplication allele was further scrutinized by sequencing. An SMN1-specific 1,025-bp fragment between exons 7 and 8 was amplified from five different AJ individuals with the major duplication allele using primer SMN1-E7F, which includes c.840C at the 3′ end, and primer SMN1-E8R (see Supplementary Figure S2A online).
Two sequence variants in the SMN1 gene were identified, g.27134T>G in intron 7 and g.27706_27707delAT in exon 8 (see Supplementary Figure S2B online). Further sequence analysis indicated that both polymorphisms were present in all individuals carrying the major duplication haplotype (2-5-6-10) or partial haplotype (2-5-6) and were absent from all other individuals. Using a combination of LongRange PCR and TA-cloning analysis, the presence of the two polymorphisms within the same clone in the 12 tested duplication cases (2 + 1) indicated that these polymorphisms resided within the same SMN1 allele.
Restriction fragment length polymorphism analysis for the g.27134T>G polymorphism
The g.27134T>G polymorphism resulted in the gain of an HpyCH4III restriction site (see Supplementary Figure S2C online). Restriction fragment length polymorphism (RFLP) analysis of 315 AJ control individuals revealed that none had this restriction site, indicating that the polymorphism was specific to the major AJ duplication allele. Of note, individual no. 70, with four SMN1 copies and no SMN2 copies (see Supplementary Table S5 online), was homozygous for the 2-5-6-10 allele combination but was heterozygous for the restriction site. This is consistent with the hypothesis that polymorphisms are present in only one copy of the SMN1 duplication. By RFLP analysis, a total of 49 of the 99 duplication (2 + 1) individuals were found to carry the g.27134T>G polymorphism. Therefore, testing for this polymorphism in the AJ population would identify about half of the silent (2 + 0) carriers, increasing the detection rate from 90 to 94%. Moreover, the residual risk of being an SMA carrier after a negative screening result would decrease from ~1 in 345 to ~1 in 580 ( Table 1 ).
To determine whether g.27134T>G was also present on SMN1 duplication alleles in other ethnic groups, the MLPA and RFLP analyses were performed on genomic DNA from 276 AA, 250 Asian, 262 Hispanic, and 458 CA individuals. The results indicated that g.27134T>G was present on duplication alleles in all populations examined but is only specific to the duplication alleles in Asians ( Table 3 ). However, the majority (81%) of AA individuals with duplications (2 + 1) were heterozygous for g.27134T>G, whereas only 21% of individuals with two copies of SMN1 were positive for this polymorphism. Of note, one of the five AA deletion carriers was also positive for the RFLP, indicating that it was present on the wild-type allele. Of note, 4 of 90 AA individuals with SMN1 duplications positive for g.27134T>G were homozygous for the RFLP, indicating that all SMN1 genes in both duplication alleles and wild-type allele carried the variant ( Table 3 ). The RFLP was present in about half of duplications (2 + 1) in Hispanic individuals, but it was also present in 5.5% of wild-type alleles. In the CA population, the RFLP was present in 15% of individuals with duplications (2 + 1) and only 0.5% of wild-type alleles (1 + 1). Conversely, in the Asian population, the RFLP was not found in individuals with two copies of SMN1 but was present in ~14% of the duplication (2 + 1) individuals ( Table 3 ). Sequencing was performed on individuals positive for the RFLP to confirm that both g.27134T>G and g.27706_27707delAT variants were present. In all cases examined, the two polymorphisms occurred together within the same SMN1 allele.
Targeted next-generation sequencing of SMN1
To further investigate SMN1 sequence variants present within the major AJ duplication allele and in the AA population, sequencing of the SMN1 gene genomic region by next-generation sequencing was performed on a selected group of samples with homozygous deletion of SMN2. The 32.7 kb target region included the entire SMN1 gene (NG008691.1) and part of the upstream and downstream sequences. Sequencing of the region for 12 samples was performed on an Illumina GAII sequencer. The sequences were analyzed using NextGENE software (SoftGenetics). The average sequence coverage was 297 reads per base pair, and those variants with reads >5% and total coverage ≥20× were called. Variants were grouped and compared based on SMN1 copy number, the presence of g.27134T>G, and ethnic group. Only variations specific to each group were further investigated.
Four additional novel variants, g.11678G>T, g.15774G>A, g.22804G>A, and g.26190A>G, were identified in both AJ and AA individuals carrying the g.27134T>G polymorphism ( Table 4 ). All four variants were validated by Sanger sequencing and were further confirmed in an additional 10 samples from each group (data not shown). These variants occurred concomitantly with g.27134T>G and g.27706_27707delAT and, therefore, delineated a conserved haplotype on duplication alleles in the AJ population and on both duplications and wild-type alleles in the AA population. Among the AA samples sequenced, no other sequence variants were identified that would distinguish the duplication alleles from the wild-type alleles.
Discussion
The American College of Medical Genetics and Genomics has recommended offering carrier screening for SMA to all couples regardless of race or ethnicity.18,26 However, because most mutations involve copy-number loss, carrier detection is limited by the false-negative rate, which varies from 4 to 27% based on ethnicity due to the inability to detect silent (2 + 0) carriers with two copies of SMN1 on one chromosome 5 and deletion on the other.1,17 To help overcome this limitation and improve carrier detection, founder alleles with either deletions or duplications of SMN1 carrying unique polymorphisms were sought to distinguish the silent (2 + 0) carriers from wild-type (1 + 1) individuals, which is not possible using current methods. The AJ population was initially selected for the haplotype studies because previous founder mutations have been identified in shared haplotype blocks, which has facilitated gene discovery for recessive disorders prevalent among AJ individuals.19,20,27 The feasibility of this approach is further supported by a recent study of an SMN1 deletion founder allele in the US Hutterite population that identified a shared haplotype and a single origin for mutations in the population, with an estimated SMA carrier frequency of one in eight.28
To search for potential founder alleles, microsatellite analyses were performed with markers flanking the SMN1 locus, and marker trait association studies were performed with four of the most tightly linked markers (D5S681-D5S435-MS1-D5S610). Because SMN1 and SMN2 are located within a segmental duplication, markers were chosen that were outside of the repeat region to eliminate potential confounding results. Comparison of the haplotype frequencies in deletion (1 + 0) and duplication (2 + 1) individuals with those in AJ controls identified two deletion- and three duplication-specific haplotypes with P ≤ 0.001 ( Table 2 ). Of these haplotypes, one was present in ~46.4% (32 of 69) (see Supplementary Table S5 online) of the duplication alleles tested but was absent in all 78 AJ controls, making it highly specific for the duplication allele and a candidate for further study (see Supplementary Table S5).
Sequencing 1,025 bp between SMN1 intron 7 and exon 8 in individuals carrying the major duplication haplotype identified two tightly linked variants, g.27134T>G in intron 7 and g.27706_27707delAT in exon 8 (see Supplementary Figure S2A,B online). The g.27134T>G transversion introduced an HpyCH4III restriction site (see Supplementary Figure S2C), which was present in the major duplication (2 + 1) carriers but not in any of the 315 AJ controls. Thus, an AJ individual with two copies of SMN1, who is positive for g.27134T>G and/or g.27706_27707delAT, would be considered an SMA silent (2 + 0) carrier. Detection of these variants would increase SMA carrier detection rate in the AJ population from 90 to 94% and reduce the residual risk after a negative carrier screening result from ~1 in 345 to 1 in 580 ( Table 1 ). Similarly in the Asian population, an individual found to have two copies of SMN1 will have his/her residual risk of being an SMA carrier reduced from 1 in 628 to 1 in 702, if negative for the two variants. However, if positive, those individuals would be identified as silent (2 + 0) carriers for SMA. For all potential silent (2 + 0) carriers, regardless of whether they carry g.27134T>G, further testing of their parents will confirm carrier status because one parent should have at least three copies of SMN1 and the other should have one copy. In addition, analysis of multiple generations will provide confirmation through phased haplotype associations.
Moreover, the two linked polymorphisms were also present at different frequencies in the AA, Hispanic, and CA populations. In these populations, g.27134T>G and g.27706_27707delAT were more frequent among duplication (2 + 1) carriers than those with one or two SMN1 copies. Although these variants were not specific to the duplication alleles, residual risk estimates could be modified in all three populations after screening negative or positive for these polymorphisms ( Table 4 ). The effect was most dramatic in the AA population, which has the lowest SMA carrier detection rate (71%) due to the high prevalence of duplication alleles.1 The residual risk increases from 1 in 121 to 1 in 34 if an AA individual with two copies of SMN1 is positive for g.27134T>G and/or g.27706_27707delAT and decreases to 1 in 396 if negative ( Table 1 ). The CA population screening data for the g.27134T>G allele shown in Table 3 indicates a different allele frequency distribution than that identified in the AJ population and is in agreement with published studies of SMN1 copy number in North American CAs.1
Additional studies in AJ and AA individuals of the entire SMN1 genomic region using a next-generation sequencing strategy identified four novel intronic sequence variants that in conjunction with g.27134T>G and g.27706_27707delAT form a conserved single-nucleotide polymorphism haplotype across the SMN1 gene, indicating that it represents an ancient haplotype specific to AJ duplication alleles and enriched in AA duplication alleles ( Table 4 ). Although a negative result for the single-nucleotide polymorphism haplotype does not rule out the possibility that an individual is a silent carrier, it reduces the residual risk in all populations examined. Additional next-generation sequencing of AJ individuals with the minor duplication haplotypes, not detected using g.27134T>G, may help to identify diagnostic polymorphisms that would further improve detection rates.
Disclosure
L.E. and R.J.D. are named inventors on a pending patent application for spinal muscular atrophy filed by Mount Sinai School of Medicine. The other authors declare no conflict of interest.
References
Hendrickson BC, Donohoe C, Akmaev VR, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet 2009;46:641–644.
Pearn J . Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 1978;15:409–413.
Meldrum C, Scott C, Swoboda KJ . Spinal muscular atrophy genetic counseling access and genetic knowledge: parents’ perspectives. J Child Neurol 2007;22:1019–1026.
Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165.
Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999;64:1340–1356.
Wirth B, Schmidt T, Hahnen E, et al. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997;61:1102–1111.
Chen KL, Wang YL, Rennert H, et al. Duplications and de novo deletions of the SMNt gene demonstrated by fluorescence-based carrier testing for spinal muscular atrophy. Am J Med Genet 1999;85:463–469.
Echaniz-Laguna A, Miniou P, Bartholdi D, Melki J . The promoters of the survival motor neuron gene (SMN) and its copy (SMNc) share common regulatory elements. Am J Hum Genet 1999;64:1365–1370.
Monani UR, McPherson JD, Burghes AH . Promoter analysis of the human centromeric and telomeric survival motor neuron genes (SMNC and SMNT). Biochim Biophys Acta 1999;1445:330–336.
Lorson CL, Hahnen E, Androphy EJ, Wirth B . A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 1999;96:6307–6311.
Cartegni L, Krainer AR . Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002;30:377–384.
Mailman MD, Hemingway T, Darsey RL, et al. Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome. Hum Genet 2001;108:109–115.
Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ . Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A 2004;130A:307–310.
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368.
Huang CH, Chang YY, Chen CH, et al. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet Med 2007;9:241–248.
Anhuf D, Eggermann T, Rudnik-Schöneborn S, Zerres K . Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum Mutat 2003;22:74–78.
Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 2002;4:20–26.
Prior TW ; Professional Practice and Guidelines Committee. Carrier screening for spinal muscular atrophy. Genet Med 2008;10:840–842.
Risch N, Tang H, Katzenstein H, Ekstein J . Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet 2003;72:812–822.
Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R . Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat 2010;31:1240–1250.
Arkblad EL, Darin N, Berg K, et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord 2006;16:830–838.
Scheffer H, Cobben JM, Matthijs G, Wirth B . Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur J Hum Genet 2001;9:484–491.
Ogino S, Leonard DG, Rennert H, Wilson RB . Spinal muscular atrophy genetic testing experience at an academic medical center. J Mol Diagn 2002;4:53–58.
Inoue K, Lupski JR . Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet 2002;3:199–242.
Ogino S, Gao S, Leonard DG, Paessler M, Wilson RB . Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Genet 2003;11:723.
ACOG Committee on Genetics. ACOG committee opinion no. 432: spinal muscular atrophy. Obstet Gynecol 2009;113:1194–1196.
Diaz GA, Gelb BD, Risch N, et al. Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations. Am J Hum Genet 2000;66:1821–1832.
Chong JX, Oktay AA, Dai Z, Swoboda KJ, Prior TW, Ober C . A common spinal muscular atrophy deletion mutation is present on a single founder haplotype in the US Hutterites. Eur J Hum Genet 2011;19:1045–1051.
Acknowledgements
Sequencing was performed at the Genomics Core Facility, Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine. This work was supported in part by a National Center for Research Resources grant (UL1RR029887) for the Mount Sinai Institutes for Clinical and Translational Sciences, from the National Institutes of Health; and a Biochemical/Molecular Genetics Fellowship from the Genzyme Corporation (to M.L. and L.L.). The authors thank Tracy Brandt for her help in next-generation sequencing.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure S1
(PDF 96 kb)
Supplementary Figure S2
(PDF 808 kb)
Supplementary Table S1
(PDF 22 kb)
Supplementary Table S2
(PDF 28 kb)
Supplementary Table S3
(PDF 23 kb)
Supplementary Table S4
(XLS 27 kb)
Supplementary Table S5
(XLS 53 kb)
Supplementary Methods and Procedures
(PDF 54 kb)
Rights and permissions
About this article
Cite this article
Luo, M., Liu, L., Peter, I. et al. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet Med 16, 149–156 (2014). https://doi.org/10.1038/gim.2013.84
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.84
Keywords
This article is cited by
-
Population WGS-based spinal muscular atrophy carrier screening in a cohort of 1076 healthy Polish individuals
Journal of Applied Genetics (2023)
-
Development and validation of a 4-color multiplexing spinal muscular atrophy (SMA) genotyping assay on a novel integrated digital PCR instrument
Scientific Reports (2020)
-
SMN1 gene copy number analysis for spinal muscular atrophy (SMA) in a Turkish cohort by CODE-SEQ technology, an integrated solution for detection of SMN1 and SMN2 copy numbers and the “2+0” genotype
Neurological Sciences (2020)
-
Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors
European Journal of Human Genetics (2019)
-
Spinal Muscular Atrophy (SMA) in the Therapeutic Era
Current Genetic Medicine Reports (2019)
