
Abstract
Purpose:
The aim of this study was to explore the clinical management of incidental findings. Advances in the speed and sensitivity of genetic technologies have not only improved the diagnostic rate but also result in an increase in unanticipated diagnoses. Recent debate on such “incidental findings” has considered whether or not to actively search for and, then, disclose incidental findings. In our experience, many incidental findings need to be investigated in family members before their clinical significance can be assessed. This adds complexity to the debate about disclosure.
Methods:
Using anonymized clinical examples, we illustrate the downstream implications when a result reveals an incidental abnormality of potential clinical significance that is not related to the reasons for doing the test.
Results:
Our examples illustrate that the determination of clinical significance may require participation of family members in both testing and surveillance.
Conclusion:
The need to investigate multiple relatives in order to decide whether or not a finding is clinically significant has implications for consent and disclosure practices. Communication with, and care for, relatives who have no reason to suspect particular diagnoses is a challenge for any health-care service. These costs also need to be taken into account as genetic testing enters mainstream medicine.
Genet Med 15 11, 896–899.
Similar content being viewed by others
Main
The last few years have seen genetic diagnostic tools move from targeted gene analysis to genome-wide approaches. One such genomic approach, array comparative genome hybridization (aCGH), has already entered routine clinical practice and has replaced karyotyping as the first-line investigation for children with developmental delay.1 Whole-exome and whole-genome sequencing are also rapidly finding a place in clinical services. What these techniques have in common is that they have the ability to identify pathogenic mutations that were not previously detectable, and thus, diagnostic rates have improved. However, as with any more-sensitive test, these techniques are also more likely to find unanticipated results: results that are apparently unrelated to the presenting problem but independently require clinical management.2,3 Such results can provide clear cut additional diagnoses but, given the novelty of many, may also be of uncertain clinical significance. One way to determine pathogenicity is by tracking the genetic finding with clinical features in a family. A variety of terms has been used to describe such findings: “unexpected”, “unanticipated”, “unsollicited”, “secondary” or “nonpertinent”. Here, we use “incidental findings” (IFs) to reflect their apparently incidental nature to the clinical question that led to the test.4,5 Our clinical experience is that IFs rarely have clear-cut clinical significance at the point of discovery. Rather, they are potential IFs whose significance may only become clear after a series of clinical investigations, both of the patient as well as their (possibly unsuspecting) relatives.
Here, we present two illustrative examples in which aCGH testing revealed an IF of potential clinical significance. Detailed laboratory and clinical investigations were required to establish the pathogenicity of this finding. These two examples highlight that consent and disclosure practices need to take this uncertainty, as well as the potential familial implications, into account. Much of the literature has focused on whether or not to disclose uncertain IFs, but we argue that this should not be seen as a simple choice if family investigations are required. The rapidly reducing cost of genome technologies has been used as a justification for their entry into clinical practice, but the additional investigations in several individuals to determine clinical relevance has not been included in such deliberations.
Clinical Example 1
Sophie (not real name), aged 4 years had array comparative genome hybridization requested by a pediatric neurologist because of developmental delay. When the result showed a chromosome 3p25.3 duplication encompassing exons 2 and 3 of the von Hippel–Lindau (VHL) gene, she was referred to the clinical genetic service for assessment.
Clinical Example 2
Two siblings Sarah (not real name) and Peter (not real name) were seen by the Clinical Genetics service in the past because of a history of seizures and developmental delay. Their karyotypes were normal at that time, and an undiagnosed recessive condition was suspected. Peter subsequently died at the age of 13 years due to a presumed seizure. Sarah was re-referred in her 20s, and aCGH testing demonstrated the same duplication as in example 1 (3p25.3).
This duplication was not thought to be the cause of the presenting features; however, a duplication of a known tumor-suppressor gene was considered to be a potential IF.
Von Hippel–Lindau disease
Mutations in the VHL gene cause this eponymous condition. Characteristic features include brain and spinal hemangioblastomas, retinal angiomas, renal cysts and tumors, pancreatic cysts, phaeochromocytomas, and other less common manifestations. More than 95% of individuals who have a mutation in the VHL gene will have developed at least one feature of the condition by 65 years of age, and 86% by the age of 40 years.6 In both the cases, neither clinical features nor the family history were suggestive of VHL. However, some VHL features may only be detectable radiologically or biochemically many years before clinical presentation, so further investigations were warranted to establish whether the children or their families were at risk of the condition.
Clinical Management
Example 1: Testing of Sophie’s parents revealed that her mother Ann (not real name) (aged 38 years) had the same duplication, but her maternal grandmother did not. Testing of her maternal grandfather was not possible, so we could not determine if the duplication was de novo in Ann or paternally inherited. This information was relevant because the presence of the duplication in an unaffected person who had reached old age would indicate that it was unlikely to be disease causing.
Example 2: There was no stored DNA from Peter (deceased) to check if he had the same duplication as his sister, Sarah, but it was shown to be present in their mother Mary (aged 60 years). Mary attended one appointment to receive her result but has declined any further follow-up surveillance appointments. Whether this is because of a recent bereavement she has suffered, or because she has not understood the potential significance, is not clear. To date, Mary has not reported any clinical symptoms suggestive of VHL. Further molecular work (fluorescent in situ hybridization and multiplex ligation-dependent probe amplification) on the duplication did not clarify its pathogenicity.
This particular duplication had not been previously described and although pathogenicity was uncertain, we assumed that VHL gene function might be affected. International surveillance recommendations (based on consensus expert opinion) suggest a minimum of: annual eye examination, 24-h urine collections, and abdominal and brain/spinal cord imaging at varying ages.7 Retinal angiomas are usually the first manifestation of VHL, so screening for this starts at 5 years of age, while brain and abdominal imaging start in early adulthood.
Sophie and Ann had five different surveillance tests, but no VHL features were found. We noted that both the families struggled to understand this potential IF and were anxious about investigations recommended for apparently healthy individuals as a result of investigations for seizures or developmental delay. We consider that this presents a significant extra burden for families and health services, yet this has not been adequately highlighted in the optimism surrounding genomic medicine. Given the penetrance of VHL mutations, at least one feature of the condition would be expected by now, at least in Mary.6 Although we can be cautiously optimistic that this potential IF is not clinically significant, a decision must now be made to see whether yet more surveillance is indicated over the next few years.
Discussion
Although the attendant-improved diagnostic yields of new technologies are to be welcomed, the discovery of findings apparently incidental to the presenting features, but whose pathogenicity are uncertain, will require careful clinical management. The UK government recently announced its plans to sequence the entire genomes of 100,000 National Health Service patients, and these downstream clinical and ethical implications will need careful consideration.8 The American College of Medical Genetics and Genomics recently issued recommendations on the reporting of IFs from clinical exome and genome sequencing. This advocates that laboratories seek and report specific mutations regardless of the clinical reason for doing the test.9 The report does not address the downstream implications of requiring family studies to help determine the clinical significance of a potential IF. We argue that the following issues require consideration:
Consent for genetic testing
Good clinical care includes, among other things, providing patients with sufficient information to make decisions about the investigations or interventions they pursue. Achieving a balance between sufficient information and information overload can be a challenge, particularly for tests that can reveal an almost infinite number of possible results. Individuals need to understand what new genetic tests can reveal, as well as the possibility of uncertainty about their results. Furthermore, they need to understand that investigations of their relatives may be important in the ascertainment of pathogenicity, and the complexities around contact will need clarification. This is set against a background of media coverage and public understanding that often portrays genetics as clear-cut and highly determinative.10 Many publications highlight the importance of discussing IFs during the consent process but do not mention the downstream follow-up or familial investigations that may be required.11,12 Evidence suggests that the possibility of an IF is rarely explored in any detail at the time of testing. Seeking the cooperation of relatives for further investigation, which might reveal health risks for people who have not sought health advice, can be difficult.13
Disclosure of results
As the pace and scale of genetic testing increases, there is less time to prepare patients for the result of any genetic test. An IF may fall outside the expertise of the clinician who ordered the test, and referral to others may be necessary. This process, with its associated time delays and numbers of appointments involved, is likely to add to the anxiety of families already burdened with new unexpected genetic information and unlinks consent and disclosure practices.
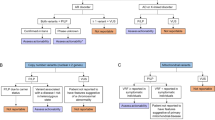
Recent debate about disclosure of IFs, both in clinical and research settings, has not yet reached a consensus.12,13,14,15,16 Options suggested range from full disclosure of all results to disclosing only those with known clinical significance, and/or which have an intervention that can prevent, treat, or mitigate disease. What our examples highlight is that it may be extremely difficult to withhold details of IFs, even if a conclusion is that they are not clinically significant, because further investigations of the patient, and their relatives, may be required to come to this conclusion. This is summarized in Figure 1 .

Illustration of the steps in delineating an incidental finding of potential clinical significance.
Familial implications
A family history of a particular condition usually means that relatives will have some awareness that they too might be at risk but, if something is found incidentally, this might suggest that there is no awareness of the suspected condition in the family. For a potential IF, family members may need to be alerted and investigated before they know whether the finding is important for them. Lack of any family history is likely to make the need for such cascade screening more difficult to understand. Furthermore, clinicians may be uncertain what, if any, duties they have to warn relatives about risks when the risks are only clarified after the relative has been contacted. The appropriate contact of relatives, while minimizing their anxiety, is a challenge which will need to be addressed as genomic investigations, often pitched as “personalized” medicine, finds susceptibility to disease that may be shared by family members. To what extent clinicians need to pursue investigations of relatives where intrafamilial communication does not happen, particularly in examples such as this where the risks are uncertain, will need to be clarified.
Long-term follow-up
Even if the pathogenicity of an IF is confirmed, the onset of symptoms may not be for many years, thus raising questions about long-term follow-up. Current health-care systems are not well equipped to deal with the recording of familial information, future risks to health, nor of managing several family members. As new and relevant information emerges, a robust mechanism to identify, recontact, and review family members is required.
Lessons from research
The potential discovery of clinically relevant yet unexpected or incidental information is also an issue in the research context. Research governance usually requires clear up front policies about such matters, and many studies have decided not to report IFs back to participants.15,17 Such clear policies are more difficult in clinical practice since the relationship between researcher and participant is different to that between clinician and patient. The researcher, bound by rules derived from the Declaration of Helsinki, undertakes researches for which the participant has given clear consent18; the clinician must also include considerations of patient welfare in such decisions. A clinically significant finding (especially those where an intervention is available) may be disclosed in the clinic, even though an equivalent research result might not be. Even so, some large research studies, for example, UK and US Biobanks, have acknowledged the need for qualified disclosure of IFs in some settings.19,20
Conclusion
The clinical impact of IFs may perversely be greater when the clinical significance of the finding is initially unclear. Further investigations in several relatives may be required before the significance, both for the patient and the relatives, can be ascertained. Once ascertained, surveillance may not be necessary for many years, so careful follow-up arrangements will need to be made. Simply framing disclosure issues in terms of “to tell or not to tell” will not cover the complexities surrounding IFs.
Although genomic technologies will often provide clear-cut individual diagnoses, we welcome a wider debate on the management of IFs, of potential or uncertain clinical significance, that incorporates good consent and disclosure practices, long-term follow-up arrangements, and appropriate communication with relatives.
Disclosure
The authors declare no conflict of interest.
References
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764.
Pichert G, Mohammed SN, Ahn JW, Ogilvie CM, Izatt L . Unexpected findings in cancer predisposition genes detected by array comparative genomic hybridisation: what are the issues? J Med Genet 2011;48:535–539.
Hogan J, Turner A, Tucker K, Warwick L . Unintended diagnosis of Von Hippel Lindau syndrome using Array Comparative Genomic Hybridization (CGH): counseling challenges arising from unexpected information. J Genet Couns 2013;22:22–26.
Kohane IS, Masys DR, Altman RB . The incidentalome: a threat to genomic medicine. JAMA 2006;296:212–215.
vanZwieten M, Willems DL, Litjens LL, Schuring-Blom HG, Leschot N . How unexpected are unexpected findings in prenatal cytogenetic diagnosis? A literature review. Eur J Obstet Gynecol Reprod Biol 2004;120:15–21.
Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med 1990;77:1151–1163.
Maher ER, Neumann HP, Richard S . von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011;19:617–623.
UKGovernment. DNA tests to fight cancer. 2013; http://www.number10.gov.uk/news/dna-tests-to-fight-cancer/. Accessed 8 March 2013.
Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. American College of Medical Genetics and Genomics: Genet Med 2013;15:565–574.
Mail D . Faulty gene dramatically increases risk of breast cancer in MEN Daily Mail 2010; http://www.dailymail.co.uk/health/article-1292401/Faulty-gene-dramatically-increases-risk-breast-cancer-MEN.html Accessed 8 March 2013.
PHG Foundation. Next Steps in the Sequence: The Implications of Whole Genome Sequencing for Health in the UK. Cambridge: PHG Foundation, 2011.
Christenhusz GM, Devriendt K, Dierickx K . To tell or not to tell? A systematic review of ethical reflections in incidental findings arising in genetics contexts. Eur J Hum Genet 2013;21:248–255.
Downing NR, Williams JK, Daack-Hirsch S, Driessnack M, Simon CM . Genetics specialists’ perspectives on disclosure of genomic incidental findings in the clinical setting. Patient Educ Couns 2013;90:133–138.
Townsend A, Adam S, Birch PH, Lohn Z, Rousseau F, Friedman JM . “I want to know what’s in Pandora’s Box”: comparing stakeholder perspectives on incidental findings in clinical whole genomic sequencing. Am J Med Genet A 2012;158A:2519–2525.
Bredenoord AL, Kroes HY, Cuppen E, Parker M, van Delden JJ . Disclosure of individual genetic data to research participants: the debate reconsidered. Trends Genet 2011;27:41–47.
Wolf S, Lawrenz FP, Nelson CA, et al. Managing incidental findings in human subjects research: analysis and recommendations. J Law Med Ethics 2008;36:219–248, 211.
Simon C, Shinkunas LA, Brandt D, Williams JK . Individual genetic and genomic research results and the tradition of informed consent: exploring US review board guidance. J Med Ethics 2012;38:417–422.
World Medical Association. WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects. 1964; http://www.wma.net/en/30publications/10policies/b3/index.html. Accessed 12 March 2013, 2013.
Knoppers BM, Kharaboyan L . “Deconstructing” Biobank Communication of Results. SCRIPTed. 2009;6:677–684.
Wolf SM, Crock BN, Van Ness B, et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet Med 2012;14:361–384.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Crawford, G., Foulds, N., Fenwick, A. et al. Genetic medicine and incidental findings: it is more complicated than deciding whether to disclose or not. Genet Med 15, 896–899 (2013). https://doi.org/10.1038/gim.2013.165
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.165
Keywords
This article is cited by
-
CGH analysis in Colombian patients: findings of 1374 arrays in a seven-year study
Molecular Cytogenetics (2018)
-
Incidental or secondary findings: an integrative and patient-inclusive approach to the current debate
European Journal of Human Genetics (2018)
-
Non-invasive prenatal testing for aneuploidy and beyond: challenges of responsible innovation in prenatal screening
European Journal of Human Genetics (2015)
