
Abstract
Purpose:
The primary objective of this study was to determine the safety of idursulfase in Hunter syndrome patients aged 5 years or younger.
Methods:
Idursulfase (0.5 mg/kg) was administered intravenously on a weekly basis (52 infusions per patient) in an open-label study. Safety monitoring included adverse events, anti-idursulfase antibodies, vital signs, physical examination, 12-lead electrocardiogram, concomitant medications or procedures, and laboratory testing (clinical chemistry, hematology, and urinalysis). The following exploratory efficacy outcomes were assessed at baseline and at weeks 18 or 36 or 53: urinary glycosaminoglycan levels, liver or spleen size, developmental milestones, and growth indices. Pharmacokinetic parameters were assessed at week 27.
Results:
Twenty-eight boys aged 1.4–7.5 years were enrolled (one discontinued for noncompliance) in the study. All the patients reported adverse events (16 patients (57%) reported possibly or probably treatment-related adverse events). The only severe adverse event was sleep apnea (two patients); others were mild or moderate. Sixteen patients had infusion-related adverse events, a similar proportion as previously reported. Thirteen patients (46%) experienced at least one serious adverse event: pyrexia and bronchopneumonia were the most common (three patients each). No clinically important drug-related changes in laboratory parameters or vital signs or electrocardiograms were reported. Nineteen patients (68%) developed anti-idursulfase immunoglobulin G antibodies. Growth rates remained within normal age-related ranges. Developmental quotients were lower than normal but remained stable. By week 18, organ size and urinary glycosaminoglycan levels decreased as compared with baseline and remained stable throughout the study.
Conclusion:
Idursulfase safety, tolerability, and efficacy were similar to that previously reported in males ≥5 years.
Genet Med 16 6, 435–441.
Similar content being viewed by others
Introduction
Hunter syndrome, or mucopolysaccharidosis II, is a rare (prevalence 1 in 162,000 male live births),1 X-linked, and recessive disease caused by a deficiency or absence of the enzyme iduronate-2-sulfatase.2 Iduronate-2-sulfatase, a lysosomal storage enzyme, is required for the degradation of glycosaminoglycans (GAG). When the enzyme is deficient, toxic levels of the GAGs such as heparan sulfate and dermatan sulfate accumulate in the lysosomes, causing the cells to enlarge, leading to organ failure, tissue dysfunction, and reduced life expectancy.3
The consequence of GAG accumulation is a progressive multiorgan disease that demonstrates great heterogeneity among patients, both in age of onset and severity and in the organs affected.4 Death usually occurs in the second or third decade of life, most often from respiratory and/or cardiac failure.3 The two usually considered distinct subtypes of Hunter syndrome are (i) attenuated (or mild) and (ii) severe. With either type, patients appear normal at birth. Patients with the severe phenotype begin to develop the signs between 2 and 4 years of age with neurological and cognitive impairments appearing before the age of 6 years.3 Patients with the attenuated subtype may not show signs until late childhood or early adolescence.5 There is, however, no demonstrated biochemical difference between the two types, and, in practice and from the clinical point of view, the severe and attenuated phenotypes are the two ends of a spectrum of clinical severity.
A recombinant form of human iduronate-2-sulfatase (idursulfase, Elaprase; Shire Human Genetic Therapies, Lexington, MA) has been approved in over 50 countries for enzyme replacement therapy of Hunter syndrome. The recommended dosage of idursulfase is 0.5 mg/kg of body weight, administered once a week as an intravenous (i.v.) infusion over 1–3 h.6
The safety and efficacy of idursulfase were evaluated in a randomized, double-blind, placebo-controlled, phase III study in 96 patients with Hunter syndrome (NCT00069641).7 In an open-label extension of the pivotal study (NCT00630747), effects of long-term idursulfase treatment were evaluated in enrolled patients for up to 3 years.8 These past studies, however, included only patients aged 5 years and older. As younger patients were not included in the previous studies, this new study intended to examine the safety and efficacy of idursulfase in patients aged 5 years or younger. In addition, the study was designed to provide a basis for evaluating the pharmacokinetic profile and pharmacodynamic effects of both single and repeat doses of idursulfase.
The primary objective of this phase IV, open-label study was to evaluate the safety and tolerability of idursulfase, administered to male patients with Hunter syndrome aged 5 years or younger. The following two secondary objectives were also evaluated: (i) the pharmacodynamic effects of idursulfase in these patients as measured by urinary GAG (uGAG) levels and (ii) idursulfase single- and repeat-dose pharmacokinetic parameters in the study patients at weeks 1 and 27, respectively. Several exploratory objectives were evaluated including the following: (i) changes in liver and spleen volumes by abdominal ultrasound, (ii) assessment of routine development milestones using the Denver developmental screening test II (Denver II),9 and (iii) growth indices (height, weight, and head circumference).
Materials and Methods
The study was conducted in compliance with the International Conference on Harmonization, Good Clinical Practice guidelines, and the US Food and Drug Administration Institutional Review Board regulations in the 21 Code of Federal Regulations 56.10 The protocol, protocol amendments, and patient informed consent were reviewed and approved by the Institutional Review Board/Independent Ethics Committee at each participating institution. The ClinicalTrials.gov identifier number is NCT00607386.
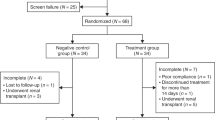
The study comprised three periods and a follow-up phase. A screening period of 14 days was followed by a 3-day baseline period when baseline assessments were taken. The treatment period lasted 52 weeks with a total of 52 infusions administered per patient. The first infusion of idursulfase could immediately follow the completion of the baseline assessments and must have been completed not more than 24 h thereafter. This was considered week 1. An end-of-study visit occurred during week 53. A follow-up phone call or clinic visit occurred 30 days after the last infusion ( Figure 1 ). The study was monitored by PharmaNet (Princeton, NJ), using local monitors in the regions where the sites were located.

Study design. aFollow-up by phone call or clinic visit.
Inclusion criteria for this study were as follows: (i) male, age ≤5 years, with a diagnosis of Hunter syndrome based on biochemical criteria either documented in the medical history or established at screening, defined as a deficiency in iduronate-2-sulfatase enzyme activity of ≤10% of the lower limit of the normal range as measured in plasma, fibroblasts, or leukocytes (based on the normal range of the measuring laboratory); and (ii) a normal enzyme-activity level of at least 1 other sulfatase measured in plasma, fibroblasts, or leukocytes (based on the normal range of the measuring laboratory). Patients were excluded if they had received treatment with another investigational therapy within 30 days prior to enrollment or had previously received idursulfase. Patients were also not allowed to enroll if they had a medical condition that would make the implementation of the protocol difficult, such as a tracheotomy, or if they had known hypersensitivity to any of the components of idursulfase.
Idursulfase dosing
Dosing was calculated based on the patient’s weight (0.5 mg/kg) at each visit. Patients received their first treatment at week 1 and every week thereafter for 1 year up to a maximum of 52 infusions for the duration of the study. The drug was administered as a continuous i.v. infusion over a minimum of 3 h. Patients were discharged from the clinic ~5 h after completion of the infusion at weeks 1 and 27 to accommodate for pharmacokinetic sample collection. On other infusion days, patients were discharged from the clinic ~2 h after completion of the infusion at weeks 2 and 3 and 1 h after completion of the infusion at weeks 4 through to 52.
Assessments and analyses
Idursulfase safety was evaluated through the assessment of treatment-emergent adverse events (AEs), concomitant medications and surgical procedures, vital signs, physical examinations, clinical laboratory testing (clinical chemistry, hematology, and urinalysis), 12-lead electrocardiograms, and monitoring of anti-idursulfase antibodies. All AEs were monitored in accordance with International Conference on Harmonization Good Clinical Practice guidelines to ensure the safety of patients. The relationship of each AEs to the study drug was recorded using the following three categories: not related, possibly related, and probably related to study drug (as assessed by the study investigators). The severity was recorded on a four-degree scale: mild, moderate, severe, and life threatening. A serious AE was defined as any AE occurring at any dose that results in death, life-threatening AE, hospitalization, persistent or significant disability/incapacity, or congenital anomaly or birth defect. An infusion-related AE was defined as an AE that (i) began either during or within 24 h after the start of the infusion and (ii) was judged as possibly or probably related to the study drug. The patient’s anti-idursulfase antibody status was determined using a multitiered approach at the Shire Bioanalytical and Biomarker Development Laboratory as per European Medicines Agency and US Food and Drug Administration guidelines on immunogenicity testing.11,12 All the confirmed positive samples were tested in vitro for activity-neutralizing antibody and cell-based internalization–neutralizing antibody activity. A sample was considered overall positive for neutralizing antibodies if found positive for activity-neutralizing or cell-based internalization–neutralizing antibodies.
Secondary efficacy end points were the mean change from baseline to week 52 in uGAG levels (normalized for microgram of GAG per milligram of creatinine) and single-dose and repeat-dose pharmacokinetic parameters at weeks 1 and 27, respectively. Exploratory variables included the following: liver and spleen measurements by abdominal ultrasound at baseline and at weeks 18, 36, and 53; routine developmental milestones using Denver II at baseline and at weeks 18, 36, and 53; and growth indices (height, weight, and head circumference recorded at baseline and at weeks 18, 36, and 53).
Analysis of uGAGs was conducted at the Shire Bioanalytical and Biomarker Development Laboratory and normalized to urine creatinine. The method used was based on the adaptation to 96-well plates of the dimethylmethylene blue dye–binding assay of De Jong et al.13 After internal validation, Shire adopted the uGAG reference ranges of the Mayo Medical Laboratories, which uses the same dimethylmethylene blue analysis method.14 The previously used normal reference range for uGAGs was developed for children aged 5 years or older and was not applicable for this study population.15
Liver size was assessed by abdominal ultrasound examination. Ultrasound was used rather than magnetic resonance imaging due to the young age of the patients; to minimize cooperation requirements; and to avoid the need for general anesthesia or sedation, which pose particular risks to patients with mucopolysaccharidosis II. Given that ultrasound cannot generate reliable estimates of liver volume, a validated index of liver size (ILS) by Dittrich et al.16 was used in this analysis. The craniocaudal liver extension (height) was measured in the following three standardized section planes: the section level of the liver in the anterior axillary line, the medioclavicular line, and the perpendicular section plane in the sternal line. These three standardized measurements were then used to estimate the ILS using the formula published by Dittrich et al.16

For measurement of spleen volume, after adjustment to give the largest section area in the longitudinal and transverse plane, the maximum extent of the spleen was measured three times as length (L), depth (DL), or as breadth (B) and depth (DB). The ultrasound measurements and the volume of the spleen were calculated as per the methods and the formula for the volume of an ellipsoid by Dittrich et al.16

Routine developmental milestones were assessed using the Denver II.9 The developmental domains assessed were: personal/social, adaptive fine motor, language, and gross motor. To put the results of the Denver II into context, quotients (the ratio of observed scores to expected scores for age) were calculated for each domain. The observed developmental ages in each of the four general areas were determined by the investigator as the midpoint of the age range identified by the failure of three consecutive tasks. The overall mean of the Denver II developmental quotient was calculated as the average of the four developmental domain quotients.
Statistical methods
This was an open-label study and not designed for formal hypothesis testing. All statistical analyses were descriptive and were performed using SAS software (SAS Institute, Cary, NC). The proposed sample size was 30 patients, representing a similar size to treatment groups in the placebo-controlled registration trials. The sample size was determined outside of statistical considerations.
Results
Baseline demographics and clinical characteristics are shown in Table 1 . Twenty-eight patients were enrolled in the study. Of the 28 patients, 1 (3.6%) discontinued the study after 4 weeks of treatment due to lack of compliance. All 28 patients were included in the safety analysis population. The mean age at entry was 4.0 years, with a range of 1.4–7.5 years. Exemptions to enter the study were granted to four patients aged older than 5 years (6.5, 7.5, 6.3, and 6.2 years). These patients fulfilled all other enrollment criteria. They had been identified at ≤5 years of age, but their study-site initiation was delayed due to logistical issues in study start-up. For one patient (6.5 years of age), confirmation of the diagnosis by biochemical criteria was only obtained prior to study entry when he was older than 5 years. The mean age at diagnosis was 3.5 years (range: 0.2–6.5 years). The mean weight at baseline was 20.0 kg (range: 11.8–28.6 kg); the mean height was 101.9 cm (range: 79.7–119.9 cm); and the mean head circumference was 53.3 cm (range: 48.6–57.2 cm).
All 28 patients took at least 1 concomitant medication during the study. Painkillers, antipyretics, antibiotics, and respiratory treatments were used to treat infections and respiratory disease that are common in Hunter syndrome. In addition, antipyretic and antiallergy medications (such as corticosteroids) were used to prevent or treat infusion-related AEs.
Treatment compliance and exposure to study drug
Patients received a total of 1,401 successfully administered idursulfase infusions out of a potential total of 1,404 (99.8%). Idursulfase was administered (0.5 mg/kg i.v.) over a minimum of 3 h. Of the 28 patients, 25 (89.3%) did not miss any infusions during the course of the study. Three of the 28 patients (10.7%) missed 1 infusion. Nine (32.1%) had one to three partial infusions during the study, mostly due to loss of vein access or infusion-related reactions. The mean duration of partial infusions was 2.3 h, ranging from 0.6 to 3.3 h. Mean duration of exposure to the study drug was 347.2 days or 49.6 weeks.
Primary outcomes: safety and tolerability
The incidence and severity of AEs are summarized in Table 2 . All 28 patients experienced at least 1 AE, and all but 2 AEs were mild or moderate in severity. Of the 28 patients, 2 (7.1%) experienced severe AEs. In both of these patients, severe AEs were sleep apnea, which did not cause interruption of the study drug, and the patients were treated with continuous positive airway pressure. The investigator considered both of the severe AEs unrelated to idursulfase treatment. Thirteen of 28 patients (46.4%) experienced at least 1 serious AE; however, no life-threatening AEs were reported.
The most commonly reported treatment-emergent AEs among the 28 patients were the following: pyrexia (n = 25 (89.2%)), upper respiratory tract infection (n = 18 (64.3%)), cough (n = 16 (57.1%)), respiratory tract infections (n = 12 (42.9%)), rhinitis (n = 11 (39.3%)), vomiting (n = 10 (35.7%)), and viral upper respiratory tract infection (n = 9 (32.1%)). The treatment-emergent AEs judged by each investigator to be probably or possibly related to the study treatment are summarized in Table 3 . Of 28 patients, 16 (57.1%) had at least 1 drug-related treatment-emergent AE, and an equal number experienced at least 1 infusion-related and treatment-emergent AE. The most common treatment-emergent AEs considered to be possibly or probably related to treatment were pyrexia, rash, urticaria, and vomiting. All these AEs were also judged as infusion related and are consistent with the safety profile from earlier clinical trials of idursulfase.6,7,8
No clinically important changes in laboratory parameters, vital signs, or electrocardiograms were reported. Of the 28 patients, 19 (67.9%) developed immunoglobulin G antibodies on at least 1 occasion. Fifteen (53.6%) had neutralizing antibodies on at least 1 occasion during the study.
Secondary clinical outcomes
Urinary GAG. Mean (±SD) baseline-normalized uGAG levels were 738.3 (±165.2) µg/mg creatinine (range: 431.5–1050.9 µg/mg). Decreases in uGAG levels were evident in all age groups and appeared as early as the week 18 time point and continued through to week 53. A mean (±SD) decrease from baseline in uGAG levels of 368.0 (±165.4) µg/mg creatinine (range: −794.5 to −109.6 µg/mg) was observed at week 18, and a mean (±SD) decrease of 400.3 (±180.3) µg/mg creatinine (range: −784.0 to −80.2 µg/mg) was observed at week 36. At week 53, the mean (±SD) decrease from baseline in uGAGs was 402.4 (±162.1) µg/mg creatinine (range: −750.3 to −104.3 µg/mg), representing a mean decrease of 54.4% (range: −11.7 to −81.8%; Figure 2 ). None of the patients were normalized at the end of the study (range: 1.1–3.6 times the upper limit of normal for age at week 53).

Mean percent change from baseline in index of liver size, spleen volume, and urinary GAGs over 53 weeks. Liver size estimated with the use of validated index of liver size and ultrasound measures of three standard imaging sections. GAG, glycosaminoglycan.
Liver size. Mean (±SD) baseline ILS was 118.9 (±30.9) cm2 (range: 67–226 cm2). A comparison to estimated age-specific nomograms for ILS is possible, and all patients had larger-than-normal ILS at baseline (mean value was ~1.8-fold the upper limit of normal of reference children). At week 18, the mean (±SD) ILS had decreased 25.4 (±22.0) cm2 (range: −78.7 to +21.7 cm2) as compared with baseline. At week 36, the mean (±SD) decrease was 25.1 (±19.0) cm2 (range: −72.9 to +3.3 cm2); at week 53, the mean (±SD) decrease was 23.2 (±30.0) cm2 (range: −64.3 to +47.0 cm2), representing a mean decrease of 17.4% (range: −47.8 to +48.4%) as compared with baseline.
Spleen volume. Mean (±SD) spleen volume at baseline was 160.0 (±56.6) ml (range: 75.2–309.3 ml). All patients had larger-than-normal spleen volumes at baseline (mean value was ~2.2-fold the upper limit of normal of reference children). By week 18, spleen volume decreased in the overall population and at all subsequent time points as compared with baseline. At week 18, mean (±SD) spleen volume had decreased by 41.4 (±36.1) ml (range: −120.0 to +36.4 ml); at week 36, the mean (±SD) decrease was 33.9 (±39.1) ml (range: −135.3 to +44.4 ml); and at week 53, the mean (±SD) decrease was 36.4 (±41.2) ml (range: −114.8 to +57.9 ml). The 53-week mean decrease was 20.6% (range: −55.1 to +37.2%) as compared with baseline.
Growth indices. The mean (±SD) head circumference of 53.3 (±2.2) cm (range: 48.6–57.2 cm) at baseline was within the normal range for all age groups. At week 53, the overall mean (±SD) head circumference had increased slightly to 54.1 (±2.2) cm, reflecting a slight increase in circumference in each age category. Centers for Disease Control and Prevention head circumference growth charts include data only from 0 to 3 years of age because head growth slows down after this period, and pediatricians do not typically assess head circumference after 3 years of age. Only four patients in this study were aged 3 years or younger at both baseline and week 53. Due to the small number of patients who were aged 3 years or younger at both baseline and week 53, the effect of idursulfase on head circumference could not be assessed fully.
Patients experienced normal growth as assessed by mean increases from baseline in height and body weight. According to the Centers for Disease Control and Prevention growth charts for boys, the annualized growth rates were similar to those expected in these age groups.17 The rates of growth for patients in each category at week 53 were the following: 8.1 cm/year for patients aged <2 years, 5.4 cm/year for patients aged ≥2 to <5 years, and 5.3 cm/year for patients aged ≥5 years. The SD of the annualized growth rate for patients aged <2 years was 6.86 cm, for patients aged ≥2 to <5 years was 4.22 cm, and for patients aged ≥5 years was 4.35 cm. The overall SD for patients aged <5 years was 4.58 cm.
Denver developmental screening test II. At baseline, the overall mean Denver II quotients across age groups and across the four domains were <0.85. Only patients in the ≥2 and <5 years of age category had a normal gross motor mean quotient. The quotients of the other three domains (personal or social, adaptive fine motor, and language) and for the other age categories were below normal. After baseline, overall changes to Denver II quotients were small, and at week 53, patients did not demonstrate a consistent upward or downward trend across any of the scales. At week 53, there was a mean increase of 2.9% from baseline in the Denver II quotient in personal or social development. However, there was a mean decrease from baseline in adaptive fine motor skills (0.6% decrease), language skills (2.3% decrease), and gross motor skills (7.0% decrease).
Pharmacokinetics. Serum concentrations of idursulfase increased rapidly for the first 60–90 min after the start of infusion, reaching a plateau or continuing to rise more slowly for the remaining 3-h of infusion. Maximum serum concentration (Cmax) was attained usually at the end of the infusion at 180 min at weeks 1 and 27. After the end of infusion, idursulfase serum concentrations declined rapidly with a mean (±SD) terminal half-life (t1/2) of 160 (±69) min (n = 26) and 138 (±24) min (n = 11, antibody-negative patients) at weeks 1 and 27, respectively. Mean (±SD) t1/2 was 64 (±19) min for the 7 antibody-positive patients with measurable concentration profiles at week 27. In this study, key pharmacokinetic parameters of systemic exposure (Cmax, area under the concentration–time curve extrapolated to infinity) and clearance (and apparent volume of distribution of steady state) for idursulfase were comparable to those previously reported in the pivotal trial.7 These pharmacokinetic parameters were comparable across the different weight (<20, 20 to <30, 30 to <40, 40 to <50, and ≥50 kg) and age groups (<11, 11 to <18, and ≥18 years of age) in both studies. These results indicate that the total systemic exposure and clearance rate of idursulfase are not affected by age or body weight.
Discussion
The primary purpose of this open-label study was to evaluate the safety of idursulfase administered to male children aged 1.4–7.5 years with Hunter syndrome. Efficacy parameters were also measured as secondary end points. No placebo-controlled group was enrolled; therefore, the discussion and interpretation of the study results are based on the comparison of safety and available efficacy data from the pivotal study. This study shares several aspects with the earlier placebo-controlled registration study, such as treatment-naive patients with Hunter syndrome, the same weekly idursulfase dose (0.5 mg/kg), and a controlled study environment.
The main difference is that the current study population is much younger, with a mean age of 4.0 years compared with a mean age of 15.4 years in the pivotal study.7 As a result, a number of methodological choices were obligatory. The younger population made the assessments of idursulfase treatment efficacy through the 6-min walk test and forced vital capacity impractical. Instead, changes in liver and spleen sizes were used as a measure of clinical efficacy. Unlike the registration study, which used magnetic resonance imaging–derived liver or spleen volumes, the current study used an ILS developed for ultrasound. The ILS was chosen because there are no established and accurate ultrasound methods to determine liver volumes. Ultrasound-derived spleen volumes could be calculated, but differences in normal reference ranges with magnetic resonance imaging exist. This study also used age-specific uGAG reference ranges because levels in normal children aged 5 years or younger are significantly higher than that in older children and, in addition, differ between the study age groups. This contrasts with the single cutoff value used for the upper limit of normal in the registration study.7 To account for all the differences in methodology, the emphasis in the discussion of study results was kept on the relative change from baseline (rather than on the absolute change) or the proportion of patients with normal or abnormal results.
All 28 patients had at least 1 AE during the study. The most common AEs, such as pyrexia, rash, or vomiting, appeared related to the underlying disease manifestations, such as respiratory events or to infusion of the study drug. The AEs are consistent with the known safety profile of idursulfase. Most of the AEs were mild to moderate in severity, and no life-threatening AEs occurred. Overall, the safety and tolerability of idursulfase, administered 0.5 mg/kg i.v. once weekly, were similar in male patients aged 1.4–7.5 years to those observed in previous studies of male patients aged ≥5 years.7,8
The observed reductions in liver size and spleen volume in this population clearly demonstrated that idursulfase was biologically active in two major organs that are known to accumulate abnormal levels of GAG in Hunter syndrome patients. Idursulfase treatment produced a clinically meaningful decrease of ~20% from baseline in liver size and spleen volume. This is similar to the ~25% decrease in either spleen or liver volume observed in the pivotal study.6 The rapid and sustained reductions indicate that idursulfase reaches the target organs, enters the cells in active form, degrades the accumulated GAGs, and reverses much of the pathological organ enlargement in this patient population.
The analysis of uGAG levels showed the ability of idursulfase to reduce the excretion of GAGs and was therefore a biochemical indication of enzyme activity. All patients had abnormal uGAG levels at baseline: four to five times greater than the upper limit of the normal range by age group. At weeks 18 through to 53, uGAGs were reduced to 2–2.5 times greater than the upper limit of the normal range. None of the patients were normalized at the end of the study, in contrast to the results reported by Muenzer et al.7 in the pivotal study, where 16 of 32 (50%) of those receiving 0.5 mg/kg idursulfase once weekly had normalized by week 53. The magnitude of the mean percentage change ranged, however, from ~40 to 60%, which is a similar change to that observed in the idursulfase phase II/III study.
There were small changes in the developmental milestones as assessed by Denver II. As i.v. idursulfase is not expected to pass the blood–brain barrier, changes in mental function were not anticipated. The Denver II is limited to children aged ≤6 years; therefore, it was not used in the placebo-controlled registration trial, and results cannot be compared across studies. Pharmacokinetic results indicate that the total systemic exposure and clearance rate of idursulfase are not affected by age or body weight.
Conclusion
This study showed that idursulfase administered 0.5 mg/kg i.v. once weekly has a similar safety profile in male patients aged 1.4–7.5 years to that observed in previous studies of male patients aged ≥5 years. Idursulfase treatment reduced uGAGs, liver size, and spleen volume in the study population. Patient age or body weight did not affect pharmacokinetic parameters of idursulfase. The findings of this study are important as they demonstrate that idursulfase can be initiated in those patients aged younger than 5 years, to stabilize and/or improve certain somatic effects of this progressive disease.
Disclosure
R.G.: travel grants and/or speaker honoraria and/or investigator fees from Actelion, Amicus, BioMarin, Genzyme, and Shire. W.-L.H.: research grants from Shire. A.T.-S.: honoraria and travel grants from Genzyme, BioMarin, and Shire. D.A.H.W. and A.P.: employees and stockholders of Shire.
References
Meikle PJ, Hopwood JJ, Clague AE, Carey WF . Prevalence of lysosomal storage disorders. JAMA 1999;281:249–254.
Bach G, Eisenberg F Jr, Cantz M, Neufeld EF . The defect in the Hunter syndrome: deficiency of sulfoiduronate sulfatase. Proc Natl Acad Sci USA 1973;70:2134–2138.
Neufeld EF, Muenzer J . The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn, vol. 3. McGraw-Hill: New York, 2001:3421–3452.
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008;121:e377–e386.
Young ID, Harper PS, Newcombe RG, Archer IM . A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J Med Genet 1982;19:408–411.
Elaprase® (idursulfase) solution for intravenous infusion [prescribing information]. Shire Human Genetic Therapies: Cambridge, MA, 2011.
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006;8:465–473.
Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011;13:95–101.
Frankenburg WK, Dodds J, Archer P, Shapiro H, Bresnick B . The Denver II: a major revision and restandardization of the Denver Developmental Screening Test. Pediatrics 1992;89:91–97.
US Department of Health and Human Services (US Food and Drug Administration). Code of Federal Regulations––Title 21––Food and Drugs. US Food and Drug Administration. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm. Accessed 1 May 2012.
U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry Assay Development for Immunogenicity Testing of Therapeutic Proteins. U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research: Rockville, MD, 2009.
Committee for Medicinal Products for Human Use. Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins (EMEA/CHMP/BMWP/14327/2006). European Medicines Agency: London, UK, 2007.
de Jong JG, Wevers RA, Liebrand-van Sambeek R . Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidoses based on dimethylmethylene blue. Clin Chem 1992;38:803–807.
Mayo Medical Laboratories. Mucopolysaccharides (MPS) Screen, Urine. Mayo Clinic: Andover, MA, 2012. http://www.mayomedicallaboratories.com/test-catalog/Overview/84464. Accessed 28 March 2013.
Gallegos-Arreola MP, Machorro-Lazo MV, Flores-Martínez SE, et al. Urinary glycosaminoglycan excretion in healthy subjects and in patients with mucopolysaccharidoses. Arch Med Res 2000;31:505–510.
Dittrich M, Milde S, Dinkel E, Baumann W, Weitzel D . Sonographic biometry of liver and spleen size in childhood. Pediatr Radiol 1983;13:206–211.
Centers for Disease Control and Prevention. Clinical Growth Charts. Centers for Disease Control and Prevention: Atlanta, GA, 2012. http://www.cdc.gov/growthcharts/clinical_charts.htm. Accessed 15 May 2012.
Acknowledgements
This study was conducted by Shire, who also provided funding for editorial assistance to the authors. Editorial assistance was provided by Evelyn B. Kelly, PhD, and Robin Smith, PhD, of The Curry Rockefeller Group, LLC, Tarrytown, New York.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Giugliani, R., Hwu, WL., Tylki-Szymanska, A. et al. A multicenter, open-label study evaluating safety and clinical outcomes in children (1.4–7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. Genet Med 16, 435–441 (2014). https://doi.org/10.1038/gim.2013.162
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.162
Keywords
This article is cited by
-
Recent trends in mucopolysaccharidosis research
Journal of Human Genetics (2019)
-
Rare disease registries: potential applications towards impact on development of new drug treatments
Orphanet Journal of Rare Diseases (2018)
-
Clinical outcomes in idursulfase-treated patients with mucopolysaccharidosis type II: 3-year data from the hunter outcome survey (HOS)
Orphanet Journal of Rare Diseases (2017)
-
Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry
Orphanet Journal of Rare Diseases (2017)
-
Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: a retrospective observational study
Orphanet Journal of Rare Diseases (2015)
