
Abstract
Purpose: Lymphedema-distichiasis syndrome is characterized by the presence of lower limb lymphedema and supernumerary eyelashes arising from the Meibomian glands. Spinal extradural arachnoid cysts have been observed in some families but their true frequency is unknown. The aim of this study is to determine the frequency of spinal extradural arachnoid cysts in lymphedema distichiasis syndrome.
Methods: We collected clinical information from all 45 living members of a complete family of 48 members and performed molecular analysis of the FOXC2 gene in 30 individuals. We obtained spinal magnetic resonance imaging from all family members with a FOXC2 gene mutation.
Results: Twelve family members carried a mutation in the FOXC2 gene and had clinical features of lymphedema-distichiasis syndrome. Of these, 58% (seven individuals) had extradural arachnoid cysts.
Discussion: We suggest that a follow-up protocol for lymphedema-distichiasis syndrome families should include spinal magnetic resonance imaging for all affected members so that the timing of surgery for removal of these cysts can be optimized.
Similar content being viewed by others


Main
Lymphedema-distichiasis is a rare familial syndrome with autosomal dominant inheritance (OMIM 153400). It is caused by mutations in the FOXC2 gene, a forkhead family transcription factor.1 Lower limb lymphedema and distichiasis (supernumerary eyelashes arising from the Meibomian glands) characterize the syndrome. Other associated features are congenital cardiac defects, cleft palate, ptosis, and spinal extradural arachnoid cysts (SEDACs). The penetrance of the genetic defect is complete by adulthood2 but expression is variable. The frequency of SEDACs in families with lymphedema-distichiasis syndrome (LDS) has not been established, although a family was described in which 70% of members had SEDACs.3 We report on a family of 48 members over three generations in which 12 had LDS. We present data regarding prevalence of the FOXC2 mutation and the various clinical features in 30 individuals.
SUBJECTS AND METHODS
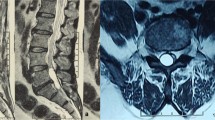
A 12-year-old asymptomatic girl attended our pediatric neurology clinic because her parents were worried that she might have spinal cysts because there was paternal family history of this malformation. Particularly, the girl'SEDACs 13-year-old cousin had recently been found to have SEDACs compressing her spine, after presenting with a gait disturbance. Our patient's grandmother had been operated on twice for SEDACs. The parents wished to ascertain whether their child suffered from a genetic defect so that any necessary measures could be taken to prevent neurological damage. We obtained a magnetic resonance imaging (MRI) scan of her spine, which showed multiple SEDACs that were not compressing the spine (Fig. 1). Physical examination revealed no neurological deficits, but we noted mild lower limb lymphedema and distichiasis. There was a paternal family history of lymphedema. In view of the suspected diagnosis of LDS, we performed molecular analysis of the FOXC2 gene in the girl. A mutation was found, and we proposed a familial study.

SEDACs (*) in the thoracic MRI of the index patient. A, Sagittal T1-weighted image shows SEDACs as well-defined hypointense structures surrounded by hyperintense normal fat in the epidural space, which is broadened. B, On a sagittal T2-weighted image with fat saturation SEDACs are hyperintense. There is no spinal cord compromise.
We collected clinical data from all living family members and performed molecular analysis of the FOXC2 gene in 30 members. Spinal MRI scans were obtained in all subjects with a FOXC2 mutation.
Molecular analysis
We obtained written informed consent from all family members studied, and parental consent for children. For adults, DNA was obtained by standard methods from ethylenediaminetetraacetic acid-anticoagulated blood samples. For children, buccal swab samples were analyzed.
In the case of the index patient, the single coding exon and splicing sites of the FOXC2 gene were analyzed by direct sequencing. Once the mutation had been identified (as Q100X), we tested for the presence of this specific mutation in other family members by means of directed polymerase chain reaction amplification and sequencing of the amplified fragment (177 bp). The primers used were
5′-CTACAGCTACATCGCGCTCA-3′ (forward) and
5′-ACCTTGACGAAGCACTCGTT-3′ (reverse).
RESULTS
The family comprises 48 individuals in three generations (Fig. 2A). We were able to obtain clinical data for all family members except for three deceased individuals: two of the first generation and individual II.14 (who died after an accident at a young age). Of the 45 living family members, 12 were symptomatic: 11 had both lymphedema and distichiasis, and subject III.9 had only distichiasis.

A, Family tree showing FOXC2 genotype and subjects with SEDACs. B, Sequencing chromatogram showing the normal sequence and the presence of the Q100X mutation in a heterozygous individual.
The clinical features of family members with the mutation and their prevalence are given in Table 1. We performed molecular analysis of the FOXC2 gene in the 12 symptomatic cases and in 18 asymptomatic family members. Only the 12 symptomatic individuals carried the mutation.
All subjects with the mutation underwent spinal MRI examination. SEDACs were found in seven of the 12 individuals with the FOXC2 mutation.
Molecular analysis
The mutation consists of a C to T transition at nucleotide number 298 of exon 1 of the FOXC2 gene (298C > T) (http://www.ensembl.org/Homo_sapiens/Transcript/Summary?db=core;t=ENST00000320354) (Fig. 2B). This DNA point mutation affects the 100th amino acid in the corresponding sequence, changing the DNA code from glutamine to a stop codon (Q100X). The same mutation has already been described in the FOXC2 gene in a lymphedema-distichiasis family.4 That the Q100X mutation is the genetic defect underlying the syndrome in the family reported here is strongly implied by the fact that it is a nonsense truncating mutation, by the mutation's segregation with the symptoms of the syndrome in this family, and by the existence of another family with the same mutation and the same syndrome.
DISCUSSION
We describe a family with a FOXC2 gene mutation and LDS, in which there was high prevalence of SEDACs. The association of primary lymphedema and distichiasis characterizes LDS. Other clinical findings have occasionally been associated with the syndrome: cleft palate, in 4-25% of cases2,4,5; ptosis, in about 30% of cases2; congenital cardiac defects, in 7-11% of cases1,2,4; and SEDACs, for which the prevalence has not been established.
In the family reported here, all individuals with the FOXC2 mutation had distichiasis. Similar findings have been reported for other LDS families and mutations.1,2,4,5 The prevalence of lymphedema in LDS is also high, but penetrance is not complete until the age of 40 years.2 In the family reported, of subjects with LDS, only one, a 13-year-old boy, did not have lymphedema. Other features such as congenital cardiac defects or cleft palate were not present in the family; however, echocardiograms were only obtained in Cases II.10, II.13, and in the index case. Four members of the family had cardiac arrhythmia, which has only been previously reported to be associated with LDS in one family.6 All of them receive antiarrhythmic drugs, and Case II.10 has undergone catheter ablation for atrial fibrillation.
Clinically, SEDACs, which represent 1% of all intraspinal tumors, usually present with gait disturbances when the cyst compresses the spinal cord, but slight neurological abnormalities can often be detected earlier.7,8 Their pathogenesis is unknown, but it is thought that the subarachnoid space communicates with the expanding cyst through a dural defect that may be congenital or caused by traumatic, inflammatory, or iatrogenic factors. The cysts expand, perhaps because their content of cerebrospinal fluid increases as a result of changes in spinal pressure.7,9 Cysts usually occur singly.7,9
Observations of SEDACs have been occasionally reported in families with LDS.3,4,8,10–12 However, the prevalence of SEDACs in LDS subjects is unclear because only one study3 has included systematic determination of their presence by spinal MRI. In the family we describe here, seven of 12 (58%) members have SEDACs; Yabuki et al.3 reported a prevalence rate of 70%. These two rates are suggestive of an association between SEDACs and LDS. Until such time as the prevalence is clarified, systematic determination of presence of SEDACs in LDS patients, especially if they have the Q100X FOXC2 mutation, is to be recommended because, although SEDACs can pass unnoticed for some time, their silent growth may produce spinal compression that needs prompt surgery if it becomes symptomatic.
To the best of our knowledge, this study is the first to suggest an association between SEDACs and the FOXC2 mutation. The same Q100X truncating mutation was found in one of the families reported by Erickson et al.,4 but SEDACs were not looked for. Yabuki et al.,3 however, determined presence of SEDACs but not the molecular nature of the mutation involved.
After 1 year of follow-up, the index case of this report is asymptomatic with regard to SEDACs, as are the three sons of Case I.5 (all of them in their forties) and Case II.15 (32 years old at the time of the study). In the family concerned, only two individuals with SEDACs developed neurological impairments, and by the time, the family came to our attention both had undergone surgery to remove the cysts. This surgery was not undertaken at the hospital of the current authors. Case I.5 has suffered neurological deficits attributable to the cysts and has been operated on twice, when she was 40 and 70 years old. Case III.13 is recovering from her second operation, but still has neurological impairment. Surgery for SEDACs usually leads to complete recovery3,7–9; possible explanations for the outcomes in the two cases of this family are that surgery was undertaken too late or was complicated by incomplete closure of the dural defect. Careful search and closure of dural defects is of extreme importance for complete resolution, as refilling of the cyst usually follows simple aspiration.9 Laminectomy is usually required, and kyphosis is a frequent postoperative complication. Therefore, although many asymptomatic patients may never develop symptoms, careful follow-up is recommended in all LDS cases.13 Although it may create anxiety in some patients, awareness of the presence of cysts is important so that surgery can be undertaken rapidly if spinal compression by SEDACs develops. MRI is the preferred technique for diagnosis and follow-up, but must be interpreted in conjunction with the findings of neurological examination.
We conclude that our findings suggest that SEDACs may be more frequent in families with LDS than previously thought, and consequently that spinal MRI should be done in all affected members. An accurate estimate of the frequency of SEDACs would help in the preparation of a follow-up protocol for the care of patients with LDS so that the timing of surgery for removal of SEDACs can be optimized thereby improving clinical outcome.
References
Fang J, Dagenais SL, Erickson RP, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 2000; 67: 1382–1388.
Brice G, Mansour S, Bell R, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet 2002; 39: 478–483.
Yabuki S, Kikuchi S, Ikegawa S . Spinal extradural arachnoid cysts associated with distichiasis and lymphedema. Am J Med Genet A 2007; 143: 884–888.
Erickson RP, Dagenais SL, Caulder MS, et al. Clinical heterogeneity in lymphoedema-distichiasis with FOXC2 truncating mutations. J Med Genet 2001; 38: 761–766.
Kumar S, Carver C, McCall S, et al. A family with lymphoedema-distichiasis where identical twins have a discordant phenotype. Clin Genet 2007; 71: 285–287.
Goldstein S, Qazi QH, Fitzgerald J, et al. Distichiasis, congenital heart defects and mixed peripheral vascular anomalies. Am J Med Genet 1985; 20: 283–294.
de Oliveira RS, Amato MC, Santos MV, Simao GN, Machado HR . Extradural arachnoid cysts in children. Childs Nerv Syst 2007; 23: 1233–1238.
Yabuki S, Kikuchi S . Multiple extradural arachnoid cysts: report of two operated cousin cases. Spine (Phila Pa 1976) 2007; 32: E585–E588.
Suryaningtyas W, Arifin M . Multiple spinal extradural arachnoid cysts occurring in a child. Case report. J Neurosurg 2007; 106( suppl 2): 158–161.
Kanaan IN, Sakati N, Otaibi F . Type I congenital multiple intraspinal extradural cysts associated with distichiasis and lymphedema syndrome. Surg Neurol 2006; 65: 162–166.
Bergland RM . Congenital intraspinal extradural cyst. Report of three cases in one family. J Neurosurg 1968; 28: 495–499.
Schwartz JF, O'Brien MS, Hoffman JC Jr, Hereditary spinal arachnoid cysts, distichiasis, and lymphedema. Ann Neurol 1980; 7: 340–343.
Ergun E, Börcek AO, Cemil B, Doğulu F, Baykaner MK . Should we operate all extradural spinal arachnoid cysts? Report of a case. Turk Neurosurg 2008; 18: 52–55.
Acknowledgements
Supported partially by Fundacion Fuentes.
We thank all the members of the family here reported for their participation in this study. We thank David Burdon for English proofreading.
Author information
Authors and Affiliations
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Sánchez-Carpintero, R., Dominguez, P., Núñez, M. et al. Spinal extradural arachnoid cysts in lymphedema-distichiasis syndrome. Genet Med 12, 532–535 (2010). https://doi.org/10.1097/GIM.0b013e3181e5c7ea
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181e5c7ea
Keywords
This article is cited by
-
Deleterious fibronectin type III-related gene variants may induce a spinal extradural arachnoid cyst: an exome sequencing study of identical twin cases
Child's Nervous System (2021)
-
Familial arachnoid cysts: a review of 35 families
Child's Nervous System (2019)
-
A novel FOXC2 mutation in spinal extradural arachnoid cyst
Human Genome Variation (2015)
