
Abstract
Alzheimer disease is the most common cause of dementia and represents a major public health problem. The neuropathologic findings of amyloid-β plaques and tau containing neurofibrillary tangles represent important molecular clues to the underlying pathogenesis. Genetic factors are well recognized, but complicated. Three rare forms of autosomal-dominant early-onset familial Alzheimer disease have been identified and are associated with mutations in amyloid precursor protein, presenilin 1, and presenilin 2 genes. The more common late-onset form of Alzheimer disease is assumed to be polygenic/multifactorial. However, thus far the only clearly identified genetic risk factor for Alzheimer disease is Apo lipoprotein E. The ε4 allele of Apo lipoprotein E influences age at onset of Alzheimer disease, but is neither necessary nor sufficient for the disease. The search continues for the discovery of additional genetic influences.
Similar content being viewed by others

Main
Clinical manifestations of Alzheimer disease
The clinical manifestation of Alzheimer disease (AD) is dementia that typically begins with subtle and poorly recognized failure of memory and slowly becomes more severe and, eventually, incapacitating. Other common findings include confusion, poor judgment, language disturbance, agitation, withdrawal, and hallucinations. Occasionally, seizures, Parkinsonian features, increased muscle tone, myoclonus, incontinence, and mutism occur.1
Death usually results from general inanition, malnutrition, and pneumonia. The typical clinical duration of the disease is 8–10 years, with a range from 1 to 25 years.
Establishing the diagnosis of Alzheimer disease
Establishing the diagnosis of Alzheimer disease relies on clinical-neuropathologic assessment.1 Neuropathologic findings on autopsy examination remain the gold standard for diagnosis of AD. The clinical diagnosis of AD (before autopsy confirmation) is correct about 80–90% of the time.2
-
Clinical signs: slowly progressive dementia
-
Neuroimaging: gross cerebral cortical atrophy3
-
Neuropathologic findings: microscopic extracellular amyloid-β (Aβ)-amyloid neuritic plaques, intraneuronal neurofibrillary tangles, and amyloid angiopathy at postmortem examination (Figs. 1 and2). The plaques should stain positively with Aβ-amyloid antibodies and negative for prion antibodies, which are diagnostic of prion diseases. The numbers of plaques and tangles must exceed those found in age-matched controls without dementia. Guidelines for the quantitative assessment of these changes exist.4,5 Aggregation of alpha-synuclein in the form of Lewy bodies may also be found in neurons in the amygdala.6
Fig 1 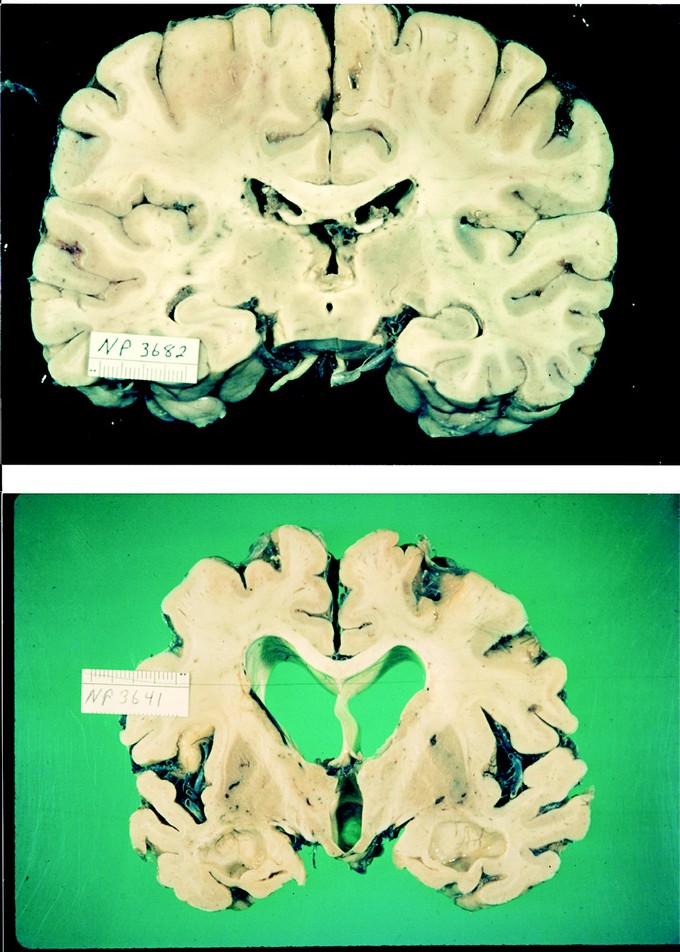
Normal adult brain (top) compared with Alzheimer brain (bottom) showing marked diffuse cortical atrophy and ventricular enlargement.
Fig 2 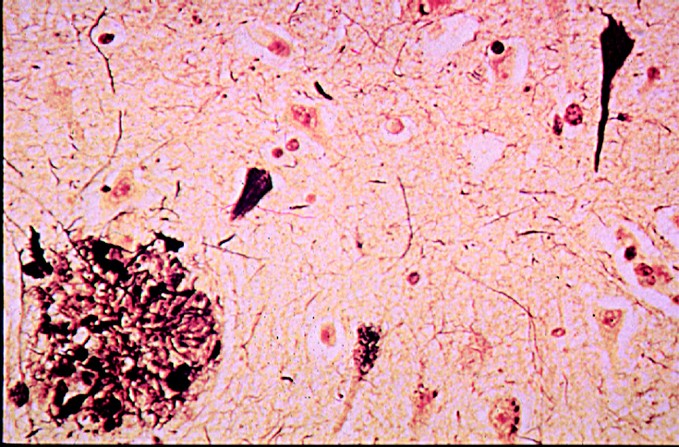
Microscopic neuropathology of Alzheimer disease showing a neuritic plaque (lower left hand corner) and neurofibrillary tangles (upper right hand corner).


Differential diagnosis of Alzheimer disease
Differential diagnosis of AD includes other causes of dementia, especially treatable forms of cognitive decline, such as depression, chronic drug intoxication, chronic central nervous system infection, thyroid disease, vitamin deficiencies (especially B12 and thiamine), central nervous system angitis, and normal-pressure hydrocephalus.1
Other degenerative disorders associated with dementia, such as frontotemporal dementia, including frontotemporal dementia with parkinsonism-17, Picks disease, Parkinson disease, diffuse Lewy body disease, Creutzfeldt-Jakob disease, and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), may also be confused with AD.7
Computerized tomography and magnetic resonance imaging are valuable for identifying some of these other causes of dementia, including neoplasms, normal-pressure hydrocephalus, frontotemporal dementia, and cerebral vascular disease.
Prevalence of Alzheimer disease
AD is the most common cause of dementia in North America and Europe, with an estimate of 4 million affected individuals in the United States.
The prevalence of AD increases with age. Mild memory loss is often called mild cognitive impairment. In many persons mild cognitive impairment is considered an early stage of AD.
The incidence of AD rises from 2.8 per 1000 person years in the 65–69 years age group to 56.1 per 1000 person years in the older than 90-year age group.8 Approximately 10% of persons older than 70 years have significant memory loss and more than half of these individuals have AD. An estimated 25–45% of persons older than 85 years have dementia.
CAUSES
About 1–6% of all AD is early onset (before age 60–65 years) and about 60% of early-onset AD is familial, with 13% seeming to be inherited in an autosomal-dominant manner (Table 1).9,10
The distinction between early-onset familial AD (EOFAD, onset before age 60–65 years) and late-onset familial AD (onset after age 60–65 years) is somewhat arbitrary. Early-onset cases can occur in families with generally late-onset disease.11
Environmental causes
No environmental agents (e.g., head trauma, viruses, toxins, low education level) have been proven to be directly involved in the pathogenesis of AD. It is often speculated that late-onset AD is the result of unknown environmental factors acting on a predisposing genetic background.12 Twin studies have implicated both genes and environment.13
Heritable causes
Chromosomal
Down syndrome.
Essentially, all persons with Down syndrome (DS) (trisomy 21) develop the neuropathologic hallmarks of AD after 40 years. More than half of individuals with DS also show, if carefully observed or tested, clinical evidence of cognitive decline.14 The presumed reason for this association is the lifelong overexpression of the APP gene on chromosome 21 encoding the amyloid precursor protein and the resultant overproduction of β-amyloid in the brains of persons who are trisomic for this gene.
The Aβ deposition in the brain may begin in the first decade of life in persons with DS.15 AD was not noted clinically or pathologically in a 78-year-old woman with partial trisomy 21, who did not have an extra copy of the APP gene.16 Two studies have found no association of Apo E genotype with age of onset of dementia in DS,17,18 but one study did find an association of onset age with a polymorphism in the APP gene.18 Schupf et al.19 found an unexplained increased risk for AD in mothers younger than 35 years, who gave birth to children with DS.
Single gene.
About 25% of AD is familial (i.e., two or more family members have AD). Familial cases seem to have the same clinical and pathologic phenotypes as nonfamilial cases (i.e., an individual with AD and no known family history of AD)20,21 and are thus distinguished only by family history or by molecular genetic testing. A large volume of research on the molecular and genetic basis of AD has been summarized by Rosenberg,22 Sleegers and van Duijn,23 Nussbaum and Ellis,24 Goedert and Spillantini,25 and Roses and Saunders.26
Late-onset familial Alzheimer disease.
Many families have multiple affected members, most or all of whom have onset of dementia after age 60 or 65 years (Table 2). Disease duration is typically 8–10 years, but ranges from 2 to 25 years. Investigations have supported the concept that late-onset AD is a complex disorder that may involve multiple susceptibility genes (reviewed and summarized by Kamboh,27 Bertram and Tanzi,28 Serretti et al.,29 and Roses and Saunders26). Bertram et al.30 have performed a meta-analysis on these data. The following information are currently available:
-
Well-documented association of late-onset familial AD (FAD) with the APOE e4 allele. The APOE e4 allele, by unknown mechanisms, seems to affect age of onset by shifting the onset toward an earlier age.31,32
-
Several other potential genes are under investigation:
-
Several other potential loci are under investigation on the following chromosomes:
-
Studies of late-onset AD in a genetically isolated Dutch population have suggested linkage of AD to markers on chromosome 1q22, 3q23, 10q22, and 11q25.57
Early-onset familial Alzheimer disease
Table 3 Early-onset familial Alzheimer disease (EOFAD): molecular genetics
-
Clinical features: EOFAD refers to families in which multiple cases of AD occur with the mean age of onset usually before age 65 years, although some studies have used age 60 or 70 years. Age of onset is usually in the 40s or early 50s, although onset in the 30s and early 60s has been reported. Campion et al.10 found a prevalence of early-onset AD in the general population of 41.2 per 100,000 persons at risk (ages 40–59 years). Sixty-one percent of these individuals with early-onset AD had a positive family history, and 13% met stringent criteria for autosomal-dominant inheritance (i.e., affected individuals in three generations). EOFAD cannot be clinically distinguished from nonfamilial AD except on the basis of family history and age of onset. The dementia phenotype is similar to that of late-onset AD, sometimes with a long prodrome.58–60
-
Molecular genetics: At least three subtypes of EOFAD (AD1, AD3, and AD4) have been identified based on the causative gene. The relative proportion of each subtype and the causative genes are summarized in Table 3.10,61–63 The APP is cleaved by alpha- and gamma-secretases to form the A beta-peptide, which is the primary component of the extracellular amyloid plaque deposited in AD (Fig. 3). Presenilin 1 (PS1) is part of the gamma-secretase complex (and PS2 is a close homolog of PS1). Thus, the three primary genes associated with EOFAD are all related to APP and A beta-amyloid molecular biology. It is likely that other genes will be identified as a cause of EOFAD because kindreds with autosomal-dominant FAD with no known mutations in presenilin 1 (PSEN1), PSEN2, or APP have been described.63,64
Table 3 Early-onset familial Alzheimer disease (EOFAD): molecular genetics Fig 3 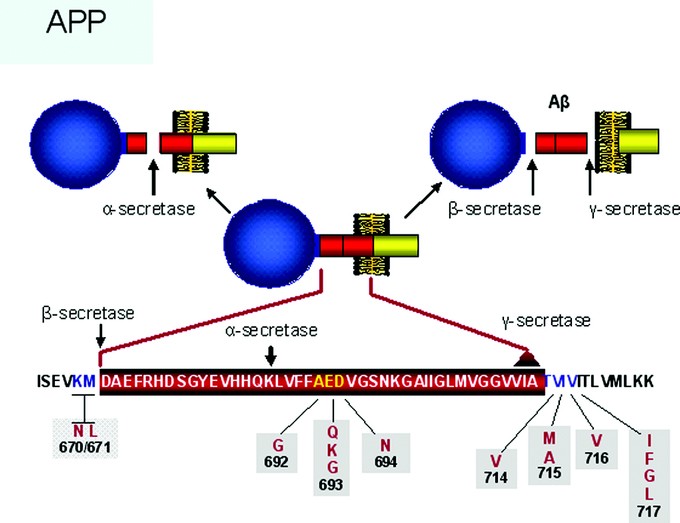
Molecular aspects of the APP gene and protein showing the sites of cleavage by α, β, and γ secretases, production of the Aβ peptide (upper right), and sites of several disease causing mutations (bottom line).

Unknown causes
Individuals with nonfamilial AD meet the diagnostic criteria for AD and have a negative family history. Onset can be anytime in adulthood. The exact pathogenesis of the disease is unknown. A common hypothesis is that nonfamilial AD is multifactorial and results from a combination of aging, genetic predisposition, and exposure to one or more environmental agents, such as head trauma, viruses, and/or toxins65 although no environmental agents have been proven to be directly involved in the pathogenesis of AD.
EVALUATION STRATEGY
Family history
A three-generation family history with close attention to the history of individuals with dementia should be obtained. For each affected individual, the age of onset of dementia should be noted. Generally, individuals with onset before age 65 years are considered to have early-onset AD and those with onset after age 65 years are considered to have late-onset AD. Medical records of affected family members, including reports of neuroimaging studies and autopsy examinations, should be obtained.
-
The diagnosis of EOFAD is made in families with multiple cases of AD in which the mean age of onset is before age 60–65 years.
-
The diagnosis of late-onset FAD is made in families with multiple cases of AD in which the mean age of onset is after age 60–65 years.
Molecular genetic testing
Late-onset familial AD
The association of one or two copies of the APOE allele e4 (i.e., genotypes e2/e4, e3/e4, e4/e4) with late-onset AD is well documented (Table 4).32,66,67
-
The association between APOEe4 and AD is greatest when the individual has a positive family history of dementia. The last column of Table 4 largely represents late-onset familial AD.
-
The strongest association between the APOE e4 allele and AD, relative to the normal control population, is with the e4/e4 genotype. That genotype occurs in about 1% of the normal control population and in nearly 19% of the familial AD population.
-
In individuals who have the clinical diagnosis of AD, the probability that AD is the correct diagnosis is increased to about 97% in the presence of the APOE e4/e4 genotype.68
-
The increased risk of AD associated with one APOE e4 allele or two APOE e4 alleles is also found in African-Americans69 and Caribbean Hispanics.70
-
Approximately 42% of persons with AD do not have an APOE e4 allele. Thus, APOE genotyping is not specific for AD. The absence of an APOE e4 allele does not rule out the diagnosis of AD2.
-
Breitner et al.71 have estimated lifetime risks for developing AD based on gender and APOE genotype (see Testing of At-risk Asymptomatic Individuals under Genetic Counseling).
The usefulness of APOE genotyping in clinical diagnosis and risk assessment remains unclear (Statements and Policies Regarding Genetic Testing).
-
Although the presence of one APOE e4 allele or two APOE e4 alleles is neither necessary nor sufficient to establish a diagnosis of AD, APOE genotyping may have an adjunct role in the diagnosis of AD because a large proportion of individuals with one APOE e4 allele or two APOE e4 alleles who are demented have been found to have neuropathologic confirmation of AD at autopsy.2,66,72,73
-
In contrast, APOE genotyping was not found to be of significant diagnostic use in identifying AD in a community-based sample with late-onset dementia.74
There is some evidence that the APOE e2 allele may have a protective effect in regard to risk for AD (Table 4).
Another way to look at this association between AD and an APOE e4 allele is with APOE e4 allele frequencies (Table 5).
Early-onset familial AD
The three known subtypes of EOFAD, called AD3, AD1, and AD474,75 can only be distinguished by molecular genetic testing (Table 3). Genetic testing of individuals who are simplex cases (i.e., a single occurrence of early-onset AD in a family) is controversial and should be undertaken in the context of formal genetic counseling.76 A small proportion (<5%) of such cases will have a mutation in PS1.
GENETIC COUNSELING
Mode of inheritance
Because AD is genetically heterogeneous, genetic counseling of persons with AD and their family members must be tailored to the information available for that family. AD is usually considered polygenic and multifactorial. EOFAD is inherited in an autosomal-dominant manner.
Risk to family members—late-onset nonfamilial Alzheimer disease
Genetic counseling for people with nonfamilial AD and their family members must be empiric and relatively nonspecific. It should be pointed out that AD is common and that the overall lifetime risk to any individual of developing dementia is approximately 10–12%.
First-degree relatives of a person with AD have a cumulative lifetime risk of developing AD of about 15–30%, which is typically reported as a 20–25% risk.77,78 This risk is about 2.5 times that of the background risk (∼27% vs. 10.4%).79,80
Disagreement exists as to whether the age of onset of the affected person changes the risk to first-degree relatives. One study found that early-onset AD increased the risk,78 whereas another study did not.77
The number of additional affected family members probably increases the risk to close relatives, but the magnitude of that increase is unclear unless the pattern in the family is characteristic of autosomal-dominant inheritance. Having two, three, or more affected family members probably raises the risk to other first-degree relatives in excess of that noted above for nonfamilial cases, although the exact magnitude of the risk is not clear. Heston et al.81 found a 35–45% risk of dementia in individuals who had a parent with AD and a sib with onset of AD before age 70 years. Jayadev et al.82 also report data suggesting that offspring of parents with conjugal AD (i.e., both parents affected) had an increased risk of dementia.
Risk to family members—early-onset familial Alzheimer disease
Many individuals diagnosed as having early-onset AD have another affected family member, although family history is negative 40% of the time.10 Family history may be “negative” because of early death of a parent, failure to recognize the disorder in family members, or, rarely, a de novo mutation. The risk to sibs depends upon the genetic status of the affected proband's parent. If one of the proband's parents has a mutant allele, then the risk to the sibs of inheriting the mutant allele is 50%. Individuals with EOFAD (and a mutation in APP, PS1, or PS2) have a 50% chance of transmitting the mutant allele to each child. The risk to other family members depends upon the status of the proband's parents. If a parent is found to be affected, his or her family members are at risk.
Related genetic counseling issues
Use of APOE genotyping for predictive testing
In contrast to the use of APOE testing as an adjunct diagnostic test in individuals with dementia, there is general agreement that APOE testing has limited value used for predictive testing for AD in asymptomatic persons. Data suggest that a young asymptomatic person with the APOE e4/e4 genotype may have an approximately 30% lifetime risk of developing AD.83 Further refinement of this risk reveals that women with an APOE e4/e4 genotype have a 45% probability of developing AD by age 73 years, whereas men have a 25% risk.71 These risks are lower—and the likely age of onset later—for persons with only one APOE e4 allele (peak age 87 years) or no APOE e4 allele (peak age 95 years). These estimates are not generally considered clinically useful; however, a research study to assess the potential use of APOE testing in relatives of individuals with late-onset AD is under way.79,84
Down syndrome
Family members of persons with DS are not at increased risk for AD.
Testing of at-risk asymptomatic EOAD family members
Testing of at-risk asymptomatic adults
Testing of asymptomatic adults at risk for EOFAD caused by mutations in the PSEN1, PSEN2, or APP gene is available clinically. Testing results for at-risk asymptomatic adults can only be interpreted after an affected family member's disease-causing mutation has been identified. It should be remembered that testing of asymptomatic at-risk individuals with nonspecific or equivocal symptoms is predictive testing, not diagnostic testing.
Preliminary results have shown that although relatively few family members choose such testing, they usually cope well with the results, which can affect personal relationships and emotional well-being.85 However, significant depression after such testing has been reported.86
Testing of at-risk individuals during childhood
Consensus holds that individuals at risk for adult-onset disorders should not have testing during childhood in the absence of symptoms. The principal arguments against testing asymptomatic individuals during childhood are that it removes their choice to know or not know this information, it raises the possibility of stigmatization within the family and in other social settings, and it could have serious educational and career implications.
Prenatal testing
Prenatal diagnosis for pregnancies at increased risk for mutations in the PSEN1 gene is possible by analysis of DNA extracted from fetal cells obtained by amniocentesis usually performed at about 15–18 weeks' gestation or chorionic villus sampling at about 10 to 12 weeks' gestation. The disease-causing allele of an affected family member must be identified before prenatal testing can be performed.
No laboratories offering molecular genetic testing for prenatal diagnosis of EOFAD caused by APP or PSEN2 mutations are listed in the GeneTests Laboratory Directory. However, prenatal testing may be available for families in which the disease-causing mutation has been identified.
Requests for prenatal diagnosis of adult-onset diseases are uncommon. Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination. Although most centers would consider decisions about prenatal testing to be the choice of the parents, careful discussion of these issues is appropriate.
Preimplantation genetic diagnosis
Preimplantation genetic diagnosis may be available for families in which the disease-causing mutation has been identified. Preimplantation diagnosis has been reported in a mother with an APP mutation.87,88
MANAGEMENT
Treatment of manifestations
The mainstay of treatment for AD is necessarily supportive and each symptom is managed on an individual basis.1 In general, affected individuals eventually require assisted living arrangements or care in a nursing home.
Although the exact biochemical basis of AD is not well understood, it is known that deficiencies of the brain cholinergic system and of other neurotransmitters are present. Drugs that increase cholinergic activity by inhibiting acetylcholinesterase produce a modest but useful behavioral or cognitive benefit in some affected individuals. The first such drug was tacrine; however, this agent is also hepatotoxic. Newer such drugs with similar pharmacologic action, such as Aricept® (donepezil),89–91 Exelon® (rivastigmine),92 and galantamine,93–95 are not hepatotoxic.
Memantine, an NMDA receptor antagonist, has shown some effectiveness in the treatment of moderate to severe AD.96–99
Antidepressant medication may improve associated depression.
Therapies under investigation
Treatment trials evaluating use of anti-inflammatory agents (NSAIDs), estrogens, nerve growth factors, ginkgo biloba, statins, beta-site cleaving enzyme (BACE) inhibitors, and antioxidants are under way or recently reviewed.100–102
Other
Vitamins and other over-the-counter medications have been used in the treatment of AD.103
Some, but not all, reports suggest that affected individuals taking 3-hydroxy-3-methylglutaryl (HMG)-coenzyme A reductase inhibitors for hypercholesteralemia have a reduced incidence of dementia.104–106
Immunization of an AD mouse model with β-amyloid has attenuated the AD pathology and stimulated the search for a possible vaccination approach to the treatment of human AD.107 A human trial of this approach was halted because of encephalitis in a few subjects.108–110 Alternative approaches to immunization therapy have been proposed.111
Thus far, treatment of symptomatic AD with estrogens has not proven beneficial.112,113
Genetics clinics
Genetics clinics are a source of information for individuals and families regarding the natural history, treatment, mode of inheritance, and genetic risks to other family members as well as information about available consumer-oriented resources.
Support groups
Support groups have been established for individuals and families to provide information, support, and contact with other affected individuals. The Resources section may include disease-specific and/or umbrella support organizations.
SUMMARY
Disease characteristics
AD is characterized by dementia that typically begins with subtle and poorly recognized failure of memory and slowly becomes more severe and, eventually, incapacitating. Other common findings include confusion, poor judgment, language disturbance, agitation, withdrawal, and hallucinations. Occasionally, seizures, Parkinsonian features, increased muscle tone, myoclonus, incontinence, and mutism occur. Death usually results from general inanition, malnutrition, and pneumonia. The typical clinical duration of the disease is 8 to 10 years, with a range from 1 to 25 years. About 25% of all AD is familial (i.e., two or more persons in a family have AD) of which about 95% is late-onset (after age 60–65 years) and 5% is early-onset (before age 65 years).
Diagnosis/testing
Establishing the diagnosis of AD relies upon clinical-neuropathologic assessment. Neuropathologic findings of extracellular β-amyloid plaques and intraneuronal neurofibrillary tangles remain the gold standard for diagnosis. The clinical diagnosis of AD, based on signs of slowly progressive dementia and findings of gross cerebral cortical atrophy on neuroimaging, is correct about 80–90% of the time. The association of the APOE e4 allele with AD is significant; however, APOE genotyping is neither fully specific nor sensitive. APOE genotyping may have an adjunct role in the diagnosis of AD in symptomatic individuals and a limited role at this time in predictive testing of asymptomatic individuals. Three forms of EOFAD caused by mutations in one of three genes (APP, PSEN1, and PSEN2) are recognized. Molecular genetic testing of the three genes is available in clinical laboratories.
Management
Treatment is supportive. Each symptom is managed on an individual basis. Assisted living arrangements or care in a nursing home is usually necessary. Drugs that increase cholinergic activity by inhibiting acetylcholinesterase produce a modest but useful behavioral or cognitive benefit in some affected individuals. Antidepressant medication may improve associated depression. An NMDA receptor antagonist is also FDA approved.
Genetic counseling
Because AD is genetically heterogeneous, genetic counseling of persons with AD and their family members must be tailored to the information available for that family. It should be pointed out that AD is common and that the overall lifetime risk for any individual of developing dementia is approximately 10–12%. Genetic counseling for people with nonfamilial AD and their family members must be empiric and relatively nonspecific. First-degree relatives of a simplex case of AD (i.e., single occurrence in a family) have a cumulative lifetime risk of developing AD of about 15–30%, which is typically reported as a 20–25% risk. This risk is about 2.5 times that of the background risk (∼27% vs. 10.4%). In contrast, EOFAD with mutations in APP, PS1 or PS2 is inherited in an autosomal-dominant manner.
RESOURCES
Alzheimer's Association National Headquarters, 225 North Michigan Avenue Fl 17, Chicago, IL 60601-7633, Phone: 800-272-3900, 312-335-8700, Fax: 312-335-1110, E-mail: info@alz.org, www.alz.org.
Alzheimer's Disease Education and Referral Center, PO Box 8250, Silver Spring, MD 20907-8250, Phone: 800-438-4380; 301-495-3334, Fax: 301-495-3334, E-mail: adear@alzheimers.org, www.alzheimers.org.
National Library of Medicine Genetics Home Reference, Alzheimer Disease.
NCBI Genes and Disease, Alzheimer Disease.
National Institute on Aging, Building 31, Room 5C27, 31 Center Drive MSC 2292, Bethesda, MD 20892, Phone: 301-496-1752, E-mail: karpf@nia.nih.gov, www.nia.nih.gov.
PUBLISHED STATEMENTS AND POLICIES REGARDING GENETIC TESTING
• American College of Medical Genetics/American Society of Human Genetics Working Group on ApoE and Alzheimer's Disease. Statement on use of apolipoprotein E testing for Alzheimer's disease. 1995.
• American Society of Human Genetics and American College of Medical Genetics. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. 1995.
• National Institute on Aging/Alzheimer's Association Working Group. Apolipoprotein E genotyping in Alzheimer's disease. Lancet 1996;347: 1091–1095.
• National Society of Genetic Counselors. Resolution on prenatal and childhood testing for adult-onset disorders. 1995.
• Post SG, Whitehouse PJ, Binstock RH, Bird TD, et al. The clinical introduction of genetic testing for Alzheimer's disease: an ethical perspective. JAMA 1997;277: 832–836.
References
Bird TD, Miller BL . Alzheimer's disease and primary dementias. In: Kasper D, Fauci A, Branwald E, Longo DL, et al., editors. Harrison's principles of internal medicine, 16th ed. New York: McGraw-Hill, 2005; 2393–2406.
Mayeux R, Saunders AM, Shea S, Mirra S, et al. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer's disease. Alzheimer's Disease Centers Consortium on Apolipoprotein E and Alzheimer's Disease. N Engl J Med 1998; 338: 506–511.
Kaye JA . Diagnostic challenges in dementia. Neurology 1998; 51: 45–52.
Braak H, Braak E . Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 1991; 82: 239–259.
National Institute on Aging-Reagan Working Group. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging 1997; 18( suppl 4): S1–S2.
Popescu A, Lippa CF, Lee VM, Trojanowski JQ . Lewy bodies in the amygdala: increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 2004; 61: 1915–1919.
Rogan S, Lippa CF . Alzheimer's disease and other dementias: a review. Am J Alzheimers Dis Other Demen 2002; 17: 11–17.
Kukull WA, Higdon R, Bowen JD, McCormick WC, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 2002; 59: 1737–1746.
Rocca WA, Hofman A, Brayne C, Breteler MM, et al. Frequency and distribution of Alzheimer's disease in Europe: a collaborative study of 1980–1990 prevalence findings. The EURODEM-Prevalence Research Group. Ann Neurol 1991; 30: 381–390.
Campion D, Dumanchin C, Hannequin D, Dubois B, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 1999; 65: 664–670.
Brickell KL, Steinbart EJ, Rumbaugh M, Payami H, et al. Early-onset Alzheimer disease in families with late-onset Alzheimer disease: a potential important subtype of familial Alzheimer disease. Arch Neurol 2006; 63: 1307–1311.
Borenstein AR, Copenhaver CI, Mortimer JA . Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord 2006; 20: 63–72.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006; 63: 168–174.
Brugge KL, Nichols SL, Salmon DP, Hill LR, et al. Cognitive impairment in adults with Down's syndrome: similarities to early cognitive changes in Alzheimer's disease. Neurology 1994; 44: 232–238.
Leverenz JB, Raskind MA . Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp Neurol 1998; 150: 296–304.
Prasher VP, Farrer MJ, Kessling AM, Fisher EM, et al. Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann Neurol 1998; 43: 380–383.
Lai F, Kammann E, Rebeck GW, Anderson A, et al. APOE genotype and gender effects on Alzheimer disease in 100 adults with Down syndrome. Neurology 1999; 53: 331–336.
Margallo-Lana M, Morris CM, Gibson AM, Tan AL, et al. Influence of the amyloid precursor protein locus on dementia in Down syndrome. Neurology 2004; 62: 1996–1998.
Schupf N, Kapell D, Nightingale B, Lee JH, et al. Specificity of the fivefold increase in AD in mothers of adults with Down syndrome. Neurology 2001; 57: 979–984.
Haupt M, Kurz A, Pollmann S, Romero B . Alzheimer's disease: identical phenotype of familial and non-familial cases. J Neurol 1992; 239: 248–250.
Nochlin D, van Belle G, Bird TD, Sumi SM . Comparison of the severity of neuropathologic changes in familial and sporadic Alzheimer's disease. Alzheimer Dis Assoc Disord 1993; 7: 212–222.
Rosenberg RN . The molecular and genetic basis of AD: the end of the beginning: the 2000 Wartenberg lecture. Neurology 2000; 54: 2045–2054.
Sleegers K, van Duijn CM . Alzheimer's disease: genes, pathogenesis and risk prediction. Community Genet 2001; 4: 197–203.
Nussbaum RL, Ellis CE . Alzheimer's disease and Parkinson's disease. N Engl J Med 2003; 348: 1356–1364.
Goedert M, Spillantini MG . A century of Alzheimer's disease. Science 2006; 314: 777–781.
Roses AD, Saunders AM . Perspective on a pathogenesis and treatment of Alzheimer's disease. Alz Dem 2006; 2: 59–70.
Kamboh MI . Molecular genetics of late-onset Alzheimer's disease. Ann Hum Genet 2004; 68: 381–404.
Bertram L, Tanzi RE . The current status of Alzheimer's disease genetics: what do we tell the patients?. Pharmacol Res 2004; 50: 385–396.
Serretti A, Artioli P, Quartesan R, De Ronchi D . Genes involved in Alzheimer's disease, a survey of possible candidates. J Alzheimers Dis 2005; 7: 331–353.
Bertram L, McQueen MB, Mullin K, Blacker D, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007; 39: 17–23.
Meyer MR, Tschanz JT, Norton MC, Welsh-Bohmer KA, et al. APOE genotype predicts when–not whether–one is predisposed to develop Alzheimer disease. Nat Genet 1998; 19: 321–322.
Khachaturian AS, Corcoran CD, Mayer LS, Zandi PP, et al. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study. Arch Gen Psychiatry 2004; 61: 518–524.
Rogaeva E, Meng Y, Lee JH, Gu Y, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 2007; 39: 168–177.
Blacker D, Wilcox MA, Laird NM, Rodes L, et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet 1998; 19: 357–360.
Dodel RC, Du Y, Bales KR, Gao F, et al. Alpha2 macroglobulin and the risk of Alzheimer's disease. Neurology 2000; 54: 438–442.
Gibson AM, Singleton AB, Smith G, Woodward R, et al. Lack of association of the alpha2-macroglobulin locus on chromosome 12 in AD. Neurology 2000; 54: 433–438.
Depboylu C, Lohmuller F, Du Y, Riemenschneider M, et al. Alpha2-macroglobulin, lipoprotein receptor-related protein and lipoprotein receptor-associated protein and the genetic risk for developing Alzheimer's disease. Neurosci Lett 2006; 400: 187–190.
Li YJ, Oliveira SA, Xu P, Martin ER, et al. Glutathione S-transferase omega-1 modifiesage-at-onset of Alzheimer disease and Parkinson disease. Hum Mol Genet 2003; 12: 3259–3267.
Reiman EM, Webster JA, Myers AJ, Hardy J, et al. GAB2 alleles modify Alzheimer's risk in APOE varepsilon4 carriers. Neuron 2007; 54: 713–720.
Pericak-Vance MA, Bass MP, Yamaoka LH, Gaskell PC, et al. Complete genomic screen in late-onset familial Alzheimer disease. Evidence for a new locus on chromosome 12. JAMA 1997; 278: 1237–1241.
Rogaeva E, Premkumar S, Song Y, Sorbi S, et al. Evidence for an Alzheimer disease susceptibility locus on chromosome 12 and for further locus heterogeneity. JAMA 1998; 280: 614–618.
Wu WS, Holmans P, Wavrant-DeVrieze F, Shears S, et al. Genetic studies on chromosome 12 in late-onset Alzheimer disease. JAMA 1998; 280: 619–622.
D'Introno A, Solfrizzi V, Colacicco AM, Capurso C, et al. Current knowledge of chromosome 12 susceptibility genes for late-onset Alzheimer's disease. Neurobiol Aging 2006; 27: 1537–1553.
Bertram L, Blacker D, Mullin K, Keeney D, et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 2000; 290: 2302–2303.
Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, et al. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science 2000; 290: 2303–2304.
Myers A, Holmans P, Marshall H, Kwon J, et al. Susceptibility locus for Alzheimer's disease on chromosome 10. Science 2000; 290: 2304–2305.
Grupe A, Li Y, Rowland C, Nowotny P, et al. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer disease. Am J Hum Genet 2006; 78: 78–88.
Riemenschneider M, Konta L, Friedrich P, Schwarz S, et al. A functional polymorphism within plasminogen activator urokinase (PLAU) is associated with Alzheimer's disease. Hum Mol Genet 2006; 15: 2446–2456.
Scott WK, Hauser ER, Schmechel DE, Welsh-Bohmer KA, et al. Ordered-subsets linkage analysis detects novel Alzheimer disease Loci on chromosomes 2q34 and 15q22. Am J Hum Genet 2003; 73: 1041–1051.
Li Y, Grupe A, Rowland C, Nowotny P, et al. DAPK1 variants are associated with Alzheimer's disease and allele-specific expression. Hum Mol Genet 2006; 15: 2560–2568.
Wijsman EM, Daw EW, Yu CE, Payami H, et al. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet 2004; 75: 398–409.
Rademakers R, Cruts M, Sleegers K, Dermaut B, et al. Linkage and association studies identify a novel locus for Alzheimer disease at 7q36 in a Dutch population-based sample. Am J Hum Genet 2005; 77: 643–652.
Bertram L, Hiltunen M, Parkinson M, Ingelsson M, et al. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med 2005; 352: 884–894.
Bensemain F, Chapuis J, Tian J, Shi J, et al. Association study of the Ubiquilin gene with Alzheimer's disease. Neurobiol Dis 2006; 22: 691–693.
Kamboh MI, Minster RL, Feingold E, DeKosky ST . Genetic association of ubiquilin with Alzheimer's disease and related quantitative measures. Mol Psychiatry 2006; 11: 273–279.
Smemo S, Nowotny P, Hinrichs AL, Kauwe JS, et al. Ubiquilin 1 polymorphisms are not associated with late-onset Alzheimer's disease. Ann Neurol 2006; 59: 21–26.
Liu F, Arias-Vasquez A, Sleegers K, Aulchnko YS, et al. A Genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet 2007; 81: 17–31.
Lampe TH, Bird TD, Nochlin D, Nemens E, et al. Phenotype of chromosome 14-linked familial Alzheimer's disease in a large kindred. Ann Neurol 1994; 36: 368–378.
Godbolt AK, Cipolotti L, Watt H, Fox NC, et al. The natural history of Alzheimer disease: a longitudinal presymptomatic and symptomatic study of a familial cohort. Arch Neurol 2004; 61: 1743–1748.
Larner AJ, Doran M . Clinical phenotypic heterogeneity of Alzheimer's disease associated with mutations of the presenilin-1 gene. J Neurol 2006; 253: 139–158.
Sherrington R, Froelich S, Sorbi S, Campion D, et al. Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet 1996; 5: 985–988.
Janssen JC, Beck JA, Campbell TA, Dickinson A, et al. Early onset familial Alzheimer's disease: mutation frequency in 31 families. Neurology 2003; 60: 235–239.
Raux G, Guyant-Marechal L, Martin C, Bou J, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. J Med Genet 2005; 42: 793–795.
Cruts M, van Duijn CM, Backhovens H, van den Broeck M, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet 1998; 7: 43–51.
Cummings JL, Vinters HV, Cole GM, Khachaturian ZS . Alzheimer's disease: etiologies, pathophysiology, cognitive reserve and treatment opportunities. Neurology 1998; 51: 2–17.
Jarvik G, Larson EB, Goddard K, Schellenberg GD, et al. Influence of apolipoprotein E genotype on the transmission of Alzheimer disease in a community-based sample. Am J Hum Genet 1996; 58: 191–200.
Martins CA, Oulhaj A, de Jager CA, Williams JH . APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology 2005; 65: 1888–1893.
Saunders AM, Hulette O, Welsh-Bohmer KA, Schmechel DE, et al. Specificity, sensitivity, and predictive value of apolipoprotein-E genotyping for sporadic Alzheimer's disease. Lancet 1996; 348: 90–93.
Green RC, Cupples LA, Go R, Benke KS, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002; 287: 329–336.
Romas SN, Santana V, Williamson J, Ciappa A, et al. Familial Alzheimer disease among Caribbean Hispanics: a reexamination of its association with APOE. Arch Neurol 2002; 59: 87–91.
Breitner JC, Wyse BW, Anthony JC, Welsh-Bohmer KA, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology 1999; 53: 321–331.
National Institute on Aging Alzheimer's Association Working Group. Apolipoprotein E genotyping in Alzheimer's disease. Lancet 1996; 347: 1091–1095.
Welsh-Bohmer KA, Gearing M, Saunders AM, Roses AD, et al. Apolipoprotein E genotypes in a neuropathological series from the Consortium to Establish a Registry for Alzheimer's Disease. Ann Neurol 1997; 42: 319–325.
Tsuang D, Larson EB, Bowen J, McCormick W, et al. The utility of apolipoprotein E genotyping in the diagnosis of Alzheimer disease in a community-based case series. Arch Neurol 1999; 56: 1489–1495.
Levy-Lahad E, Bird TD . Genetic factors in Alzheimer's disease: a review of recent advances. Ann Neurol 1996; 40: 829–840.
van der Cammen TJ, Croes EA, Dermaut B, de Jager MC . Genetic testing has no place as a routine diagnostic test in sporadic and familial cases of Alzheimer's disease. J Am Geriatr Soc 2004; 52: 2110–2113.
Farrer LA, O'Sullivan DM, Cupples LA, Growdon JH, et al. Assessment of genetic risk for Alzheimer's disease among first-degree relatives. Ann Neurol 1989; 25: 485–493.
Silverman JM, Li G, Zaccario ML, Smith CJ, et al. Patterns of risk in first-degree relatives of patients with Alzheimer's disease. Arch Gen Psychiatry 1994; 51: 577–586.
Green RC . Risk assessment for Alzheimer's disease with genetic susceptibility testing: has the moment arrived?. Alz's Care Quarterly 2002; 3: 208–214.
Cupples LA, Farrer LA, Sadovnick AD, Relkin N, et al. Estimating risk curves for first-degree relatives of patients with Alzheimer's disease: the REVEAL study. Genet Med 2004; 6: 192–196.
Heston LL, Mastri AR, Anderson VE, White J . Dementia of the Alzheimer type. Clinical genetics, natural history, and associated conditions. Arch Gen Psychiatry 1981; 38: 1085–1090.
Jayadev S . Steinbart EJ . Chi Y-Y, Kukull WA . et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol In press.
Breitner JC . APOE genotyping and Alzheimer's disease. Lancet 1996; 347: 1184–1185.
Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, et al. Genetic risk assessment for adult children of people with Alzheimer's disease: the Risk Evaluation and Education for Alzheimer's Disease (REVEAL) study. J Geriatr Psychiatry Neurol 2005; 18: 250–255.
Steinbart EJ, Smith CO, Poorkaj P, Bird TD . Impact of DNA testing for early-onset familial Alzheimer disease and frontotemporal dementia. Arch Neurol 2001; 58: 1828–1831.
Quaid KA, Murrell JR, Hake AM, Farlow MR . Presymptomatic genetic testing with an APP mutation. J Genet Couns 2000; 9: 327–345.
Towner D, Loewy RS . Ethics of preimplantation diagnosis for a woman destined to develop early-onset Alzheimer disease. JAMA 2002; 287: 1038–1040.
Verlinsky Y, Rechitsky S, Verlinsky O, Masciangelo C, et al. Preimplantation diagnosis for early-onset Alzheimer disease caused by V717L mutation. JAMA 2002; 287: 1018–1021.
Feldman H, Gauthier S, Hecker J, Vellas B, et al. Economic evaluation of donepezil in moderate to severe Alzheimer disease. Neurology 2004; 63: 644–650.
Seltzer B, Zolnouni P, Nunez M, Goldman R, et al. Efficacy of donepezil in early-stage Alzheimer disease: a randomized placebo-controlled trial. Arch Neurol 2004; 61: 1852–1856.
Petersen RC, Thomas RG, Grundman M, Bennett D, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005; 352: 2379–2388.
Rogers SL, Friedhoff LT . The efficacy and safety of donepezil in patients with Alzheimer's disease: results of a US Multicentre, Randomized, Double-Blind, Placebo-Controlled Trial. The Donepezil Study Group. Dementia 1996; 7: 293–303.
Raskind MA, Peskind ER, Wessel T, Yuan W . Galantamine in AD: A 6-month randomized, placebo-controlled trial with a 6-month extension. The Galantamine USA-1 Study Group. Neurology 2000; 54: 2261–2268.
Tariot PN, Solomon PR, Morris JC, Kershaw P, et al. A 5-month, randomized, placebo-controlled trial of galantamine in AD. The Galantamine USA-10 Study Group. Neurology 2000; 54: 2269–2276.
Mega MS, Dinov ID, Porter V, Chow G, et al. Metabolic patterns associated with the clinical response to galantamine therapy: a fludeoxyglucose f 18 positron emission tomographic study. Arch Neurol 2005; 62: 721–728.
Reisberg B, Doody R, Stoffler A, Schmitt F, et al. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 2003; 348: 1333–1341.
Tariot PN, Farlow MR, Grossberg GT, Graham SM, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291: 317–324.
Reisberg B, Doody R, Stoffler A, Schmitt F, et al. A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease. Arch Neurol 2006; 63: 49–54.
Ott BR, Blake LM, Kagan E, Resnick M, for the Memantine MEM-MD-11AB Study Group. Open label, multicenter, 28-week extension study of the safety and tolerability of memantine in patients with mild to moderate Alzheimer's disease. J Neurol 2007; 254: 351–358.
Overshott R, Burns A . Treatment of dementia. J Neurol Neurosurg Psychiatry 2005; 5( suppl 76): v53–v59.
Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J . Therapeutic approaches to Alzheimer's disease. Brain 2006; 129: 2840–2855.
Masters CL, Beyreuther K . Alzheimer's centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 2006; 129: 2823–2839.
Yaffe K, Clemons TE, McBee WL, Lindblad AS . Impact of antioxidants, zinc, and copper on cognition in the elderly: a randomized, controlled trial. Neurology 2004; 63: 1705–1707.
Wolozin B, Kellman W, Ruosseau P, Celesia GG, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57: 1439–1443.
Li G, Higdon R, Kukull WA, Peskind E, et al. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology 2004; 63: 1624–1628.
Li G, Larson EB, Sonnen JA, Shofer JB, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology 2007; 69: 878–885.
Schenk D, Barbour R, Dunn W, Gordon G, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999; 400: 173–177.
Check E . Nerve inflammation halts trial for Alzheimer drug. Nature 2002; 415: 462
Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol 2004; 14: 11–20.
Gilman S, Koller M, Black RS, Jenkins L, et al. AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005; 64: 1553–1562.
Qu B, Boyer PJ, Johnston SA, Hynan LS, et al. Abeta42 gene vaccination reduces brain amyloid plaque burden in transgenic mice. J Neurol Sci 2006; 244: 151–158.
Mulnard RA, Cotman CW, Kawas C, van Dyck CH, et al. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer's Disease Cooperative Study. JAMA 2000; 283: 1007–1015.
Wang PN, Liao SQ, Liu RS, Liu CY, et al. Effects of estrogen on cognition, mood, and cerebral blood flow in AD: a controlled study. Neurology 2000; 54: 2061–2066.
Acknowledgements
This work was supported by funding from Veterans Affairs Research Funds and NIA/NIH Grant P50 AG 005136-22.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: Dr. Bird receives licensing fees from Athena Diagnostics, Inc.
Rights and permissions
About this article
Cite this article
Bird, T. Genetic aspects of Alzheimer disease. Genet Med 10, 231–239 (2008). https://doi.org/10.1097/GIM.0b013e31816b64dc
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31816b64dc
Keywords
This article is cited by
-
Going beyond established model systems of Alzheimer’s disease: companion animals provide novel insights into the neurobiology of aging
Communications Biology (2023)
-
Biologia Futura: four questions about ageing and the future of relevant animal models
Biologia Futura (2022)
-
Rational design of novel benzisoxazole derivatives with acetylcholinesterase inhibitory and serotoninergic 5-HT4 receptors activities for the treatment of Alzheimer’s disease
Scientific Reports (2020)
-
Genome-wide analysis of genetic predisposition to Alzheimer’s disease and related sex disparities
Alzheimer's Research & Therapy (2019)
-
Subacute to chronic Alzheimer-like alterations after controlled cortical impact in human tau transgenic mice
Scientific Reports (2019)
