
Abstract
Purpose: To present the rapid-ACCE model and report our early experience of using the ACCE structure to guide systematic reviews for the rapid evaluation of emerging genetic tests.
Methods: A rapid-ACCE review uses the same 44 questions that were developed for the full-ACCE model to guide the conduct of systematic review. We combined published literature with unpublished data to estimate test performance and input from experts to help clarify qualitative issues. As questions were answered, gaps in knowledge were identified and articulated. The draft review was then sent to outside reviewers whose comments were incorporated into the final document.
Results: We conducted two reviews, both of which were completed in 6 months or less (averaging about 100 hours of primary analyst time), within modest budgets. In addition to defining the current state of knowledge about the tests, the identified gaps are expected to help define the research agendas. Both collaborating experts and study sponsors valued both the process and outcomes from the reviews.
Conclusions: Based on our early experiences, it is possible to conduct rapid systematic reviews within the ACCE structure of some emerging genetic tests to produce summaries of available evidence and identification of gaps.
Similar content being viewed by others

Main
Gene-based tests are rapidly emerging from the efforts of the Human Genome Project. As of January 2007, GeneTests listed 610 laboratories testing for 1343 genetic diseases, of which 1,046 are offered clinically.1 This reflects an annual rate of increase of about 25% between 1997 and 2006. As these tests enter the medical marketplace, clinicians, policymakers and payers must make decisions about provision or coverage of the tests and do so in a timely and affordable manner.
In the United States, evaluation of such testing falls generally into two categories: comprehensive and ad hoc. Comprehensive evaluations (e.g., U.S. Preventive Services Task Force [USPSTF], Cochrane Collaboration) are highly resource intensive and may take many months to years to complete. At present, most are done or commissioned by government entities. None of these models were developed specifically for the evaluation of screening and diagnostic tests, much less genetic tests. Moreover, the Cochrane Collaboration, in recognition of the lack of standardization of methodologies for evaluating and reporting diagnostics (as well as acknowledging the difficulties inherent to the task), formed the Standards for Reporting of Diagnostic Accuracy (STARD) Initiative in 1999.2 This work continues. Additionally, various private organizations (e.g., ECRI, Hayes Inc.) also conduct and sell technology assessment reports and occasionally address genetic tests via their standard formats. Some large payers (e.g., Blue Cross/Blue Shield, Cigna) have the resources to maintain a dedicated group to perform comprehensive technology assessments and may have proprietary methods for evaluating genetic tests. Smaller payers may apply a variety of methods, within their time and resource constraints. For our purposes, we consider all these noncomprehensive methods of evaluating genetic tests as ad hoc.
There remains a need for a standardized, rigorous format developed specifically for evaluating genetic screening and diagnostic tests that can be flexibly applied in a rapid fashion for use by different stakeholders in different test scenarios. In response to these known inadequacies, the Centers for Disease Control and Prevention (CDC) sponsored the development of the ACCE model for the evaluation of genetic tests.3,4 This model is composed of a standard set of 44 questions (see Appendix; supplemental online-only material) and builds on previously published methodologies and terminology introduced by the Secretary's Advisory Committee on Genetic Testing.5 The ACCE acronym derives from the four main domains evaluated: Analytic validity (the ability of a test to measure the genotype of interest both accurately and reliably); Clinical validity (the ability of a test to detect or predict the disorder/phenotype of interest); Clinical utility (the risks and benefits associated with the introduction of a test into practice); and Ethical, legal, and social implications (including both general issues as well as those specific to genetic tests).
With the completion of the ACCE model structure in 2004, it was adopted as the core of the UK model for evaluating genetic tests and subsequently applied to 30+ genetic tests by the fall of 2005,6 but none were published or put into the public domain. Spain has also adopted most elements of the ACCE model.7 Other countries evaluate genetic tests guided by their respective technology assessment formats. Sanderson et al.,6 in their review of their experiences with the ACCE model, have offered a number of suggestions to improve the ACCE format and have encouraged the development of “streamlined” formats, even offering a core set of issues/questions that might be addressed with every review. These suggestions may not be applicable for all stakeholders (e.g., U.S. insurers). There are continued efforts both in the United States and Europe to improve models for the evaluation of genetic tests. Despite all the activity related to developing a best model for evaluating genetic tests, the ACCE model remains the only structure that was developed specifically for this purpose, is standardized, public, and gaining acceptance and experience.
The purpose of this report is to summarize our experience using the ACCE model in a rapid fashion by combining the expertise of those who developed and used the original ACCE model (G.E.P., M.R.M.) with those with extensive experience in the use of ad hoc methods for technology assessment of emerging tests from the payer perspective (J.M.G., M.S.W.). Our combined experience provided expertise in clinical epidemiology, statistics, technology assessment, and clinical genetics. The two rapid-ACCE reviews that were performed are (1) bone morphogenetic protein receptor type II (BMPR2) mutation testing in individuals with a diagnosis of familial or idiopathic pulmonary arterial hypertension and (2) cytochrome P-450 (CYP2C9) and vitamin K epoxide reductase complex 1 (VKORC1) testing before warfarin dosing to prevent serious bleeding events.
MATERIALS AND METHODS
Both of the genetic tests/disorders reviewed by our rapid-ACCE method were emerging; thus, the evidence bases could be systematically reviewed relatively rapidly, yet still address all 44 questions. Additionally, access to gray or unpublished data from experts was made available. The core analytic team for each of the two rapid-ACCE reviews consisted of four to five investigators, with M.R.M. as the primary analyst. There were no substantive differences in the two reviews we conducted, so discussion of details to illustrate methods and results is based on only one test (BMPR2 mutation analysis).
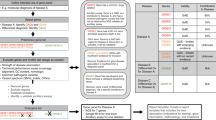
The team began the process by drafting a one-page document containing a description of the disorder, the genetic test(s), and clinical setting. This was provided to a panel of experts with clinical and research experience in the diagnosis of and testing for the disorder before a scheduled conference call. The aim of the initial teleconference was to establish agreement from the panel on the definition of the disorder and terminology, the details of the clinical setting, and application of the proposed genetic test. A preliminary conceptual flow diagram of how the test was to be used was also constructed (Fig. 1). The analyst then performed an initial review of the published literature using standard search strategies.8 Available literature was reviewed, and quantitative summaries (i.e., meta-analyses) of available data were conducted.9 In some instances, an iterative process was required to collect data. For example, a literature search might reveal that all affected individuals had the gene of interest sequenced. However, now that the major mutations have been identified, other methodologies are being employed for clinical testing. This would require the gathering of gray data from the clinical laboratories or other sources. The next step was a series of teleconferences with the panel members and other identified experts to review these summaries, to confirm any identified gaps in knowledge, and to clarify the interpretation of results. Qualitative ACCE questions were also addressed by a combination of expert opinion and review of the published and unpublished literature. Responses to all 44 questions were drafted by the analyst and reviewed by the analytic team and collaborating experts. When areas of disagreement were recognized, consensus was sought through additional discussions and data extraction (where possible), or gaps in knowledge were identified and articulated. The final draft report was then sent to outside reviewers who were knowledgeable in genetic testing, but not specifically knowledgeable about the specific disorders/tests. Their comments were addressed and included in the final report.

Flowchart of BMPR2 testing for idiopathic pulmonary artery hypertension. This flowchart, simplified for publication, summarizes the use of BMPR2 testing, at the time of the review, in the evaluation of idiopathic pulmonary artery hypertension.
RESULTS
This methodology was applied to two emerging genetic tests: (1) bone morphogenetic protein receptor type II (BMPR2) mutation testing in individuals with a diagnosis of familial or idiopathic pulmonary arterial hypertension and (2) cytochrome P-450 (CYP2C9) and vitamin K epoxide reductase complex 1 (VKORC1) testing before warfarin dosing to prevent serious bleeding events. The first review was completed in 6 months at a cost of $12,000, the second was completed in about 4 months for $23,000. The second review was actually more expensive because the funding group incurred additional expense to identify content experts and to convene a meeting to review the draft report, provide comments, and discuss the most recent research findings. Actual time spent directly working on the reviews was at least 100 hours; however, time was not recorded. Both reviews resulted in a final document, with an executive summary; the BMPR2 review was 27 pages and the CYP2C9 and VKORC1 review was 67 pages. Both reviews provided adequate information about the value and limitations of the testing and, importantly, identified gaps in evidence.
Following are results from the BMPR2 review specific to the four ACCE domains that may affect decisions about this test and are likely to be found for most emerging tests. (The results from the other review will be published in conjunction with a forthcoming ACMG clinical review). Figure 2 displays a time frame for this project. A significant portion of the six-month time period was needed to arrange mutually agreeable times for the conference calls and in discussions with the expert panel members clarifying and reaching consensus on details of clinical setting, concepts and definitions central to the reviews. The experts who contributed to this review included three pulmonologists, a pediatric cardiologist, an internist, and several laboratorians.

Time frame of the project to evaluate pulmonary arterial hypertension and BMPR2 mutation testing.
Analytic validity
There were no published data on the analytic validity for any of the laboratory methodologies that might be used to detect BMPR2 mutations. Estimates relied on unpublished data that were provided by experts. Moreover, no external proficiency programs were in place for mutation testing in these three genes. This lack of proficiency testing is not unique to the disorders in these reviews and represents a challenge for all rare disease tests.10 No data were found that address pre- or postanalytic error.
Clinical validity
Estimates of clinical sensitivity (proportion of patients with the disorder having a detectable mutation) varied among the BMPR2 mutation testing methodologies, with broad confidence intervals. Additionally, there was a lack of information about the prevalence of the mutations in the general population and their penetrance. In these instances, it was necessary to rely on expert opinion.
Clinical utility
There were no data showing that BMPR2 mutation test results affected patient care. The expert panel agreed that, at the present time, results from this testing would not influence medical care for affected individuals or unaffected family members. There are no known preventive therapies for unaffected gene carriers. Patient educational materials were evaluated and found to require a literacy level greater than that of the average adult reading level in the United States.
ELSI
Many of the issues that pertain to ELSI have been identified and discussed in other ACCE reviews.3 As pointed out by Sanderson et al.,6 this is a difficult issue to adequately address and most work has been done in the context of general principals and population screening.
DISCUSSION
These early applications of the rapid-ACCE model were deemed successful in terms of resources expended, time to deliver the final reports, and the comprehensive nature of the reviews. Furthermore, we expect that as we accumulate experience with this model, time required to complete rapid reviews will continue to decrease, with the caveat that this approach may not be applicable to certain testing scenarios.
Strengths
The clear strength of the rapid-ACCE approach is that evidence-based reviews guided by a rigorous, genetic test–specific format can be completed in a time frame that meets the decision needs and budgets of stakeholders. This is particularly important given the rapid development and dissemination of genetic tests because many of these tests will be offered commercially with limited published data, giving potential stakeholders little time to respond to requests for use and/or coverage.
Whereas a comprehensive review would generally not be used on tests with small evidence bases, we were able to apply the comprehensive, genetic test–specific ACCE structure to the small published evidence bases, allowing standard statistical methods to yield useful and timely summaries of current evidence. In addition, we added gray data and consensus opinions from experts where helpful and were able to identify gaps in evidence.
Although we have not yet done so, we also suggest that the same genetic test–specific ACCE structure and statistical methods could be applied to larger evidence/data sets in a focused way to yield sound evidence-based reviews of a limited set of ACCE questions that a stakeholder can define as key to their decisions. For example, a physician or payer could choose to initiate the review of the evidence of clinical utility. In the case of the first test that we reviewed, the lack of evidence of utility would rapidly indicate that the test is not ready for routine clinical use and review of the other categories would be unnecessary. Although ad hoc assessment methods can also use this shortcut, they are not based on a standardized public model that was developed specifically for evaluating genetic tests. The comprehensive list of 44 questions may also stimulate reviewers to seek information about aspects of a test that would otherwise not be considered.
The ACCE structure also groups questions in a way that identifies appropriate stakeholders and data sources. Test developers and laboratory personnel are more focused on analytic validity. Clinicians are interested about clinical validity and utility. Regulators and payers tend to focus on the clinical utility questions that pull all aspects of testing together in the form of decision or economic analyses. Another strength is the identification of gaps in knowledge that could be used to define research agendas. Last, the rapid- (and full-) ACCE model provides a common language upon which both reviewers and decision makers can agree, which will improve information sharing.
Limitations
The most significant limitation of the rapid-ACCE model can also be considered a strength when compared with existing methodologies. When data are limited, the rapid-ACCE approach allows the use of gray data. Gray data are considered to be a difficult area of undertaking for most evidence-based programs because such programs tend to focus solely on published literature. However, analysts must be cautious when using unpublished data due to the bias that can be introduced by data submitted by experts with a financial or intellectual investment in the use of the test.11 If experts are the only source of information, a gap in knowledge should be acknowledged. The use of multiple experts to discuss completeness and interpretations before release and then outside reviewers after preliminary release is likely to improve the reliability and reduce the risk of bias.
It can be argued that the rapid-ACCE structure has not been formally validated. This criticism applies to all evaluation methodologies in that validation implies that a gold standard exists for comparison. As noted, the ACCE model has been in use in a number of settings.
Some challenges to evidence-based reviews are not dependent on the structure or model used to organize the review. Tests with a high level of analytic and/or conceptual complexity (e.g., multiplex testing platforms, testing for complex disorders); emerging tests where evidence and experience are changing extremely rapidly; rare disorders where there will always be limited analytic and clinical data; technologies where there is no gold standard for comparison or validation (e.g., array-comparative genomic hybridization); and predictive testing where the phenotype of interest may not appear for many years may pose unique challenges for reviewers.
Last, both of the ACCE methodologies could benefit by applying a formal method of grading evidence. This element has been incorporated into our most recent review.
Lessons learned
Central to any good systematic review, even an abbreviated one, is a complete understanding of the clinical setting. Thus, we strongly encourage taking the time to thoroughly understand the nature and characteristics of the new test, its alternatives (other tests, no tests), and the clinical setting in which it is to be applied. It may be helpful to sketch out a flowchart to illustrate this. Disagreement about such details between experts can itself identify preliminary evidence gaps, as well as uncovering areas where gray data might provide benefit for the review.
Related to the above issue is the need to critically examine the genotype-phenotype association as these relationships lie at the core of the proposed utility of the testing. This begins with definition of the phenotype of interest, which may not be straightforward. Although the phenotype and genotype-phenotype relationship are frequently assumed to be self-evident, at least with regard to mendelian disorders, there is a growing body of evidence that calls this assumption into question.12–14 This will be especially true when assessing genetic tests for complex disorders, which may include some disorders formerly considered simple or mendelian. This view is supported by a 2006 guideline from Spain on the assessment of genetic testing that states that the “study of a genetic variant in the context of clinical practice is not considered (or should not be considered) until the causal relationship between that variant and a specific health problem is sufficiently established.”7 The challenges in establishing genotype-phenotype relationships in complex diseases is discussed at length in a recent series of editorials in the International Journal of Epidemiology.15
Another important concern is application of this approach to evaluating genetic tests that are already in clinical practice that often have large evidence bases (and indeterminate decision-making time frames). Many of these tests have never been systematically reviewed. Stakeholders have to decide two things: whether it is reasonable to consider removing the test from established medical practice and what will be the nature of the review (e.g., ad hoc, targeted, or comprehensive). Eddy16 has suggested that it may be a waste of resources to consider removing tests from established medical practice because “eliminating medical interventions that are already imbedded in practice is disruptive, demoralizing and difficult.” Furthermore, even good systematic reviews may be ignored, especially if reported after the test has been incorporated into practice. For example, testing for factor V Leiden and other prothrombotic mutations/factors has been available for years and continues to be used and reimbursed, despite several formal reviews suggesting the utility in most patient groups is poor and not cost-effective.3,17–19
Summary
There are a variety of stakeholders in the health care arena who must make decisions about the use of genetic tests. The principal issues for most will be the reliability of the review, the time to completion, and the resources required. Although reliability may be considered most important, the other issues can no longer be ignored. We suggest that systematic review of available evidence applied through the formal, structured and “public” ACCE model can be performed when either the evidence base is relatively small or when the stakeholders require a thorough review of only a limited set of ACCE questions (“targeted review”); to yield a reliable, evidence-based summary to inform timely decisions in an affordable manner. There may be some tests/disorders where the ACCE model, regardless of its “completeness”, may be insufficient to adequately address all key decision needs. Only further work will define whether and where the rapid-ACCE approach plays a role in future genetic test assessment.
We recognize the ongoing efforts to improve on the ACCE model, both in the United States (i.e., Evaluation of Genomic Applications in Practice and Prevention {EGAPP]) and abroad to address some of these issues. The EGAPP model will perhaps provide both structure and methods for evaluating genetic tests. Across the pond, we acknowledge the substantial efforts in the United Kingdom and Europe, under the guidance of such groups as the Genetic Testing Network Group (UK), Public Health Genomics European Network, and EuroGentest, all of which have based their work on the ACCE core principals. We eagerly await reporting of all these efforts to help with the challenge of evaluating genetic/genomic tests in a rigorous, rapid, and affordable fashion.
SUMMARY RECOMMENDATIONS FOR RAPID-ACCE REVIEWS
-
Clearly define and limit the nature of review, including matching the scope of the review to the key decisions and needs of the stakeholders.
-
Use of clinical experts (and their data) should be balanced by a thorough review of the published literature whenever possible.
-
Ask the experts to help develop a conceptual flow diagram of the testing process to ensure a detailed consensual understanding of how the testing is applied in clinical settings.
-
The following key issues should be determined before starting the review:
-
What is the disorder of interest?
-
What is the test of interest?
-
What is the clinical scenario in which the test will be applied, including the measures commonly used by clinicians to answer the primary clinical question(s) of interest?
-
-
A multipurpose test should be evaluated with respect to each of its proposed purposes
-
Enlist an experienced epidemiologist or biostatistician, preferably one with experienc in clinical diagnostics.
-
Convey gaps in evidence to interested stakeholders.
-
Read references 4, 6, and 7, at minimum, before embarking on your first rapid-ACCE review.
References
GeneTests. Available at: http://www.genetests.org/ ( accessed January 3, 2007).
Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, et al. STARD group. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. Ann Clin Biochem 2003; 40: 357–63.
National Office of public Health Genomics, CDC. ACCE model System for collecting, analyzing and disseminating information on genetic tet. Available at: http://www.cdc.gov/genomics/ACCE/fbr.htm accessed November 20, 2006.
Haddow JE, Palomaki GE, ACCE: a model process for evaluating data on emerging genetic tests. Human genome epidemiology: a scientific foundation for using genetic information to improve health and prevent disease. Khoury M, Little J, Burke W, New York Oxford University Press, 2003; 217–233.
Enhancing the oversight of genetic tests: recommendations of the SACGT. Available at: http://www4.od.nih.gov/oba/sacgt/reports/oversight_report.pdf. Accessed January 5, 2007.
Sanderson S, Zimmern R, Kroese M, Higgins J, et al. How can the evaluation of genetic tests be enhanced? Lessons learned from the ACCE framework and evaluating genetic tests in the United Kingdom. Genet Med 2005; 7: 495–500.
Márquez-Calderón S, Pérez de la Blanca EB . Framework for the assessment of genetic testing in the Andalusian Public Health System. Sevilla: Agencia de Evaluación de Tecnologías Sanitarias de Andalucía, 2006.
Sackett DL, Straus SE, Richardson WS, Rosenberg W, et al. Evidence-based medicine: how to practice and teach EBM, 2nd ed. Edinburgh: Churchill Livingstone, 2000.
Cook DJ, Mulrow CD, Haynes RB . Systematic reviews: synthesis of best evidence for clinical decisions. Ann Intern Med 1997; 126: 376–380.
Chen B, O' Connell CD, Boone DJ, Amos JA, et al. Developing a sustainable process to provide quality control materials for genetic testing. Genet Med 2005; 7: 534–549.
Roddy E, Doherty M . Guidelines for management of osteoarthritis published by the American College of Rheumatology and the European League Against Rheumatism: why are they so different?. Rheum Dis Clin North Am 2003; 29: 717–731.
Jura J, Wegrzyn P, Jura J, Koj A . Regulatory mechanisms of gene expression: complexity with elements of deterministic chaos. Acta Biochim Pol 2006; 53: 1–10.
Lanpher B, Brunetti-Pierri N, Lee B . Inborn errors of metabolism: the flux from mendelian to complex diseases. Nat Rev Genet 2006; 7: 449–460.
Dipple KM, McCabe ERB . Phenotypes of patients with “simple” mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet 2000; 66: 1729–1735.
Buchanan AV, Weiss KM, Fullerton SM . Dissecting complex disease: the quest for the Philosopher's Stone?. Int J Epidemiol 2006; 35: 562–571.
Eddy DM, How should we make decisions when we don't have good evidence? Presented at American Association of Health Plans, May 26, 2001.
Wu O, Robertson L, Twaddle S, Lowe GD, et al. Screening for thrombophilia in high-risk situations: systematic review and cost-effectiveness analysis. The Thrombosis Risk and Economic Assessment of Thrombophilia Screening (TREATS) study. Health Technol Assess 2006; 10: 1–110.
Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW . Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med 2006; 166: 729–36.
Lamberts SW, Langeveld CH, Vandenbroucke JP . Population screening for single genes that codetermine common diseases in adulthood had limited effects. J Clin Epidemiol 2006; 59: 358–364.
Acknowledgements
The authors acknowledge the expert panel that participated in the review of BMPR2 testing for idiopathic pulmonary hypertension, which was supported by the CDC through the Pulmonary Hypertension Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
A supplementary Appendix is available via the ArticlePlus feature at www.geneticsinmedicine.org. Please go to the July issue and click on the ArticlePlus link posted with the article in the Table of Contents to view this material.
Rights and permissions
About this article
Cite this article
Gudgeon, J., McClain, M., Palomaki, G. et al. Rapid ACCE: Experience with a rapid and structured approach for evaluating gene-based testing. Genet Med 9, 473–478 (2007). https://doi.org/10.1097/GIM.0b013e3180a6e9ef
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3180a6e9ef
Keywords
This article is cited by
-
Evaluating the Integration of Genomics into Cancer Screening Programmes: Challenges and Opportunities
Current Genetic Medicine Reports (2019)
-
How is genetic testing evaluated? A systematic review of the literature
European Journal of Human Genetics (2018)
-
Personalized medicine and access to health care: potential for inequitable access?
European Journal of Human Genetics (2013)
-
Study designs and methods post genome-wide association studies
Human Genetics (2012)
