
Abstract
Purpose: Routine protocols for the study of mental retardation include karyotype, analysis for fragile X syndrome, and subtelomeric rearrangements. Nevertheless, detection of cryptic rearrangements requires more sensitive techniques. Mutation screening in all known genes responsible for X-linked mental retardation is not feasible, and linkage analysis is sometimes limited. Multiplex ligation probe amplification is a recently developed technique based on the amplification of specific probes that allows relative quantification of 40 to 46 different target DNA sequences in a single reaction.
Methods: In the present study, we assessed multiplex ligation probe amplification for the detection of microduplications/microdeletions in 80 male patients with suspicion of X-linked mental retardation.
Results: We detected four copy number aberrations (5%): three duplications (GDI1, RPS6KA3, and ARHGEF6) and one deletion (OPHN1). All these changes were confirmed by other molecular techniques, and patients were clinically re-evaluated.
Conclusions: We strongly recommend the use of multiplex ligation probe amplification as a first screening method for the detection of copy number aberrations in patients with mental retardation because of its cost-effectiveness.
Similar content being viewed by others


INTRODUCTION
The molecular basis of X-linked metal retardation (XLMR) is poorly understood because of the high genetic heterogeneity of this disorder. Genes causing XLMR are thought to contribute to the specific development of human cognitive abilities. Many of these genes are conserved among vertebrates, suggesting they are engaged in basic cellular mechanisms, such as cytoskeleton organization (PAK3, OPHN1, ARHGEF6, DCX, IL1RAPL1, TM4SF2), chromatin remodeling (FMR2, MECP2, RSK2, ATRX), or signaling cascades (GDI1, PAK3, OPHN1, ARHGEF6).1,2 Mutations in most of them cause nonsyndromic mental retardation, although several are also associated with a specific syndrome.3 The search for nonspecific XLMR genes remains expensive, and findings are limited by the small number of families found to harbor mutations in the same X-linked gene. Therefore, a large number of these XLMR families remain undiagnosed in most genetics services. Routine protocols for the study of XLMR include karyotype and analysis for fragile X syndrome, which is responsible for 15% to 20% of all patients with XLMR. Thus, the genetics etiology of more than 50% of patients with mild to moderate mental retardation (MR) and 20% of patients with severe MR remains still unknown. Furthermore, detection of cryptic rearrangements requires more sensitive techniques than conventional cytogenetics. Classical polymerase chain reaction (PCR) is widely used in the study of genetic alterations, but it is not sensitive enough to detect small deletions or duplications. Recently developed technologies such as multiplex ligation probe amplification (MLPA) have become effective tools for the detection of these aberrations.4 MLPA is based in the relative quantification of specific DNA sequences by the hybridization of genomic DNA with a mixture of probes.5 This methodology allows the study of copy number changes in 40 to 46 different sequences in a single experiment. In this study, we assessed the use of MLPA in the detection of microrearrangements in patients with XLMR.
PATIENTS AND METHODS
Patients
A total of 80 unrelated male patients belonging to families from GIRMOGEN (Spanish Network for Mental Retardation Study) that met criteria for XLMR (more than one affected male family member in several generations or more than three affected brothers) were included in this study. Previous studies showed normal karyotype and no FMR1 expansions, and subtelomeric rearrangements were ruled out using MLPA for subtelomeric regions.
Methods
MLPA assay
MLPA was performed according to manufacturer's recommendations (Salsa P106, MRC, Holland). The P106 kit contains a specific probe mixture with 43 probes for 14 known genes responsible for XLMR (PQBP1, TM4SF2, ARX, IL1RAPL1, RPS6KA3, OPHN1, FACL4, DCX, PAK3, ARHGEF6, FMR1, FMR2, GDI1, and SCL6A8). Electrophoresis was performed using the ABI 310 genetic analyzer (Applied Biosystems, Foster City, CA) with Rox 500 size standard and GeneMapper software v3.0 (Applied Biosystems). GeneMapper data were exported to an Excel file to perform the analysis. Normalization of MLPA data was always required to avoid quantitative differences of measured values among samples because of variations in experimental conditions. This normalization was achieved by dividing the signal value of each probe by the sum of the signal values of all probes for each sample. Previous analysis of 20 samples from normal male subjects and 20 samples from normal female subjects allowed for normal cut-off levels to be established. A normal copy number should generate a normalized signal value of 1. For men, a deletion in a locus should result in the suppression of signal because they only have one X chromosome, whereas amplification results in an increment greater than 30%. For women, a reduction of relative signal greater than 30% was considered a deletion, and an increment in signal strength greater than 30% was considered a duplication because no control sample exceeded these values.
Assessment of MLPA sensitivity
To test the sensitivity of MLPA, we designed an experiment in which we screened a series of sample dilutions (80%, 50%, and 30%) of DNA from a 1p36 deletion carrier for subtelomeric copy number changes with Salsa PO36B (MRC, Holland).
Confirmatory studies
All MLPA findings were confirmed by other molecular techniques. The deletion affecting exons 21 and 22 of the OPHN1 gene was confirmed by PCR using specific primers: 21F (5′-TTATCATTAATCTTCCCTCTTGG-3′), 21R (5′-AAGTAGTTAGGGTCAGCTCTGG-3′), 22F (5′-AAGTTGGCCCAGGTAACTCT-3′), and 22R (5′-GTGATCCTGAAAGCATTCCT-3′).
Duplication of the RPS6KA3 gene was confirmed by using a tiling-path X-chromosome CGH array consisting of approximately 1600 genomic BAC clones derived from the X chromosome (unpublished data). Thresholds for copy numbers were established by log ratios, >0.2 for gains and ≤0.2 for losses. ARHGEF6 duplication was confirmed by fluorescence in situ hybridization (FISH) in interphase nuclei using BAC clones mapping on Xq26.3 (RP11-431K12, RP11-812C12, RP11-378N15). In the case of GDI1 duplication, we investigated its effect on mRNA expression. Total RNA of the patient and other affected male family members was extracted from whole blood. Expression of the GDI1 gene was analyzed by real time RT-PCR using TaqMan probes (Applied Biosystems). Quantification was performed against a control amplicon of the GUSB mRNA (Applied Biosystems). Furthermore, we investigated the possible duplication of the near MECP2, FLNA, and EMD genes using a specific MLPA for MECP2 (SALSA P015C; MRC) and probes for FLNA and EMD genes.
In all cases, segregation of alterations with phenotypes was confirmed by studying other members of the families.
RESULTS
Of the 80 patients, 4 (5%) analyzed using MLPA showed evidence of a cryptic aberration: three duplications (affecting RPS6KA3, GDI1, and ARHGEF6 genes) and one deletion (affecting OPHN1 gene). Pedigrees of these families are shown in Figure 1.

Pedigrees of Families 1, 2, 3, and 4. Affected individuals are represented with black symbols. Carrier women are shown with a black dot inside their symbol. The N inside symbols refers to non-affected individuals after molecular study. Empty symbols correspond to non-studied individuals.
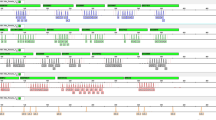
DNA copy numbers for each studied sequence were reflected by ratios between normalized peak areas and the corresponding mean of normalized peak areas of the female or male reference data set. Altered ratios for patients harboring duplications and deletions are depicted in Figure 2.

Relative peak area for each probe measured in Patient II.1 of Family 1 (A), Patient II.2 in Family 2 (B), Patient II.1 in Family 4 (C), and Patient II.1 in Family 3 (D). X-axis, 46 tested probes (for a complete list of probes, see www.mrc-holland.com). Y-axis, Normalized ratios. Bars that exceed the limit of 1.30 in A–C correspond to probes for RPS6KA3, GDI1, and ARHGEF6 genes, respectively. In D, the lack of one peak is evidence of the OPHN1 deletion (black arrow).
Family 1
The three probes for the RPS6KA3 gene were duplicated in the patient (Fig. 1A, II.1) and two affected male family members (Fig. 1A, I.5 and II.3). As expected, two women (Fig. 1A, I.2 and I.4) were diagnosed as carriers. An X-chromosome CGH array confirmed a duplication of 1Mb in Xp22.12, affecting four genes (CXorf23, FLJ14503, EIF1AX, and RPS6KA3) (data not shown). All affected male family members presented nonspecific mental retardation and robust build, and two were dyslexic.
Family 2
MLPA identified a duplication in the two probes of the GDI1 gene in the patient (Fig. 1B, II.2) and two affected uncles (Fig. 1B, I.3, I.4). As expected, the patient's mother (Fig. 1B, I.2) was diagnosed as a carrier. Expression of GDI1 was analyzed at the mRNA level, and significant differences were detected between affected male subjects and male controls with a significantly higher mRNA expression in patients carrying the duplication (from 2.8- to 5-fold compared with controls). Specifically designed MLPA showed duplication of FLNA and EMD genes, but no alterations were detected in the MECP2 gene. All affected male subjects presented moderate MR, a peculiar face with dysmorphic features, and microcephaly.
Family 3
MLPA analysis showed a deletion affecting exon 21 of the OPHN1 gene in four affected male subjects (Fig. 1C, II.1, II.4, II.6, and II.8). Using PCR, we confirmed a deletion spanning exons 21 and 22. Two women in the family (Fig. 1C, I.2 and II.7) could be diagnosed as carriers (Fig. 3). This deletion does not result in a truncated protein but includes the domain of interaction with actin, located in the carboxyl-terminal end of the protein. All affected male subjects share a conspicuous phenotype of severe MR showing strabismus, synophrys, prominent nose, and a broad high nasal root. The magnetic resonance imaging scan of two of the affected male subjects evidenced cerebellar hypoplasia and a Dandy-Walker variant malformation.

Multiplex ligation-dependent probe amplification electropherogram traces from a control sample (A), a female carrier of the OPHN1 deletion (B), and the patient with the OPHN1 deletion (C). Amplified probes are detected as fluorescent signals, the areas of which are compared and normalized to quantify the gene dosage. The lack of the peak corresponding to exon 21 of the OPHN1 gene (arrows) reveals a deletion in the patient (C). The carrier state of the mother and the sister is shown by the half-signal intensity of the peak compared with the control sample.
Family 4
MLPA showed duplication of the four probes of the ARHGEF6 gene in the patient (Fig. 1D, II.1) and his brother (Fig. 1D, II.2). Two women in the family (Fig. 1D, I.2 and I.4) were diagnosed as carriers. FISH in interphase confirmed duplication of clones RP11-812C12, RP11-378N15, and RP11-431K12 located in the region (Fig. 4). Both affected patients had moderate MR and displayed dysmorphic features.

A, Dual-color FISH used to detect the duplication in the region of the previously reported ARHGEF6 duplication. Probe: RP11-431K12 (red) and RP11-378N15 (green). B, Duplication of the RP11-812C12 clone (red arrow) in an interphase nuclei.
Assessment of multiplex ligation probe amplification sensitivity
In the series of DNA dilutions using material from an individual with a known 1p36 deletion, MLPA was able to detect alterations in samples with at least 50% of aberrant cells. Relative peak area ratios corresponding to samples with 30%, 50%, and 80% of mosaicism were 0.74, 0.6, and 0.54, respectively.
DISCUSSION
In the last few years, much interest has been focused on the identification of X-linked genes implicated in MR. OMIM lists 395 XLMR entries (syndromic and nonsyndromic), but advances in this field are limited because of the high heterogeneity of this entity. Each gene usually accounts for a small number of families; thus, families with an unknown cause clearly exceed the number of families with identified mutations.6 The development of new diagnostic tools, such as MLPA and array CGH, has facilitated the detection of deletions and duplications in patients with idiopathic MR.7,8 Some deletions and duplications are not easily detected by PCR analysis, and despite the high reliability of other methods, such as Southern blot analysis or FISH, they are laborious and not suitable for routine screening. We used an MLPA approach to detect copy number aberrations in 80 patients with XLMR, and we identified one deletion and three duplications possibly involved in MR. None of these aberrations has been described as a genomic polymorphism (see database: http://projects.tcag.ca/variation/).
Duplication of the RPS6KA3 gene seems to be disease-causing in Family 1. RPS6KA3 codes a member of the RSK family of serine/threonine kinases, acting in the Ras-mitogen–activated protein kinase (MAPK) signaling pathway.9 Activation of the RPS6KA3 protein by MAPK leads to the activation of numerous and diverse cytosolic and nuclear substrates (CREB, c-FOS, c-JUN, SRF, Nur77) and various histones, among others. RPS6KA3 protein seems to be important in chromatin-remodeling events and in gene regulation. Mutations in this gene cause Coffin-Lowry syndrome, an XLMR syndrome.10,11 Mutations causing Coffin-Lowry are heterogeneous and include truncating mutations (60% of cases) and mis-sense mutations (38% of cases). Our patient carrying the RPS6KA3 duplication does not share the typical phenotype of Coffin-Lowry syndrome, characterized by severe MR, facial dysmorphism, and progressive skeletal malformations. The other three genes that map in the duplicated region are an essential eukaryotic translation initiation factor (EIF1AX) required for the binding of the 43S complex (a 40S subunit, eIF2/GTP/Met-tRNAi and eIF3) to the 5′ end of capped RNA, an open reading frame 23 (CXorf23), and a predicted hypothetical protein (FLJ14503). We cannot rule out that other genes may contribute to the clinical phenotype. RPS6KA3 is the only known gene in this region that plays a fundamental role in neural development and signaling. We hypothesize that dosage effect of RPS6KA3 may be the major cause for the MR syndrome in this family.
Duplication of the GDI1 gene in Xq28 was detected in Family 2. This is one of the genes involved in the control of cycling between the active and inactive states of the RAB3 protein, a member of the Rab family, and has a major role in mental disorder. Mutations in GDI1 are responsible for nonspecific XLMR. Mutations described in the GDI1 gene include one non-sense and two mis-sense and are known to decrease the affinity between RAB3 and GDI1 proteins.12,13 Expression analysis of GDI1 at the mRNA level detected a significant increment in those patients carrying the duplication. Xq28 is a chromosomal region frequently affected by rearrangements and contains several genes mutated in MR. Emerin (EMD) is a serine-rich nuclear membrane protein and a member of the nuclear lamina-associated protein family. It mediates membrane anchorage to the cytoskeleton. Mutations in the EMD gene cause the X-linked recessive form of Emery Dreifuss dystrophy.14,15 Filamin A, encoded by the FLNA gene, is a widely expressed protein that regulates reorganization of the actin cytoskeleton by interacting with integrins, transmembrane receptor complexes, and second messengers. Recent reports suggest that neuropathological anomalies and clinical mental retardation seen in an affected male subject with the BPNH disorder could result from a mutation of the FLNA gene.16,17 Until now, no duplication has been found to be responsible for MR in any of these genes. However, the implication of duplications in the MECP2 region, also mapping in Xq28, has recently been described in severe MR.18 Thus, we examined our patient to determine whether the duplication expanded to the MECP2 region. Results of the MLPA did not show alterations in the MECP2 gene and allowed us to discard the implication of MECP2 in our patient's phenotype.
In our study of Patient 3, we detected an intragenic deletion in the OPHN1 gene. This gene was identified through the cloning of a balanced X-12 translocation in a girl affected with MR.19 OPHN1 encodes a Rho-GTPase–activating protein (Rho-GAP) involved in the regulation of the G-protein cycle. Mutations in this gene were initially reported to be responsible for nonspecific XLMR. However, new reports suggest that OPHN1 mutations result in a recognizable syndrome. All patients display similar neuroradiological anomalies such as cerebellar hypoplasia and ventriculomegaly.20–22 We have noted the importance of magnetic resonance imaging in male patients diagnosed with XLMR to investigate these anomalies (unpublished data).
The variation detected in Family 4 was a duplication in the ARHGEF6 gene. ARHGEF6 encodes a Rac1/Cdc42-specific guanine nucleotide exchange factor. Mutations in this gene have been implicated in XLMR. ARHGEF6 is mainly expressed in the central nervous system, in ventricular zones where neuronal progenitor cells are located and in other non-neural tissues involved in different signaling cascades in Rho-mediated signaling pathways.23,24
Errors in regulators or effectors of the Rho GTPases are a significant cause of XLMR, revealing the important role of Rho signaling in cognitive function. ARHGEF6 and OPHN1 proteins directly interact with Rho GTPases, a family of regulatory molecules that play an important role in the organization of the actin cytoskeleton. GDI1 is one of the proteins controlling the activity of the small GTPases of the Rab family in vesicle fusion and intracellular trafficking. These three proteins are crucial for neuronal morphogenesis and connectivity, and all of them participate in numerous cellular processes.25
Reports in the literature indicate that deletions in XLMR genes are more frequent than duplications. Our findings highlight the advantage of MLPA compared with other previously used techniques for the detection of duplications. The use of MLPA will allow investigators to determine the real implication of these kinds of rearrangements in patients with MR. Male subjects with duplications within the long or the short arm of the X chromosome are rare, and the phenotype associated with these duplications is usually attributed to an excess of gene dosage. One of the most recent cases described in the literature is the previously mentioned microduplication syndrome involving the MECP2 region. Other high-resolution technologies, such as array CGH, also allow the screening for copy number aberrations in patients with MR.26 Nevertheless, they are still not suitable for routine diagnosis in most molecular laboratories not only because of their high cost, but also because of the difficulty in their interpretation.
MLPA allows the study of up to 46 sequences in a single experiment with results in less than 48 hours. It only requires 20 ng of human DNA and the necessary equipment present in most molecular biology laboratories. Furthermore, it is both highly specific and sensitive, allowing the detection of mosaicisms and the diagnosis of carriers, which is a powerful tool when providing genetic counseling. Families 1 and 3 went undiagnosed for a long time; the daughters, carriers of the RPS6KA3 duplication and OPHN1 deletion, will benefit from our studies by receiving genetic counseling.
However, MLPA analysis is limited to deletions/duplications closer to the selected probe mixes in the kit, although probes can be easily designed for specific regions.
We conclude that MLPA is a powerful tool for screenings of small deletions and duplications in XLMR because of its cost-effectiveness (we detected the causative mutation in 5% of patients), and it should be included in routine diagnostic protocols for patients with XLMR.
References
Ropers HH, Hamel BC . X-linked mental retardation. Nat Rev Genet 2005; 6: 46–57.
Zechner U, Wilda M, Kehrer-Sawatzki H, Vogel W, et al. A high density of X-linked genes for general cognitive ability: a run-away process shaping human evolution?. Trends Genet 2001; 1: 697–701.
Kleefstra T, Hamel BC . X-linked mental retardation: further lumping, splitting and emerging phenotypes. Clin Genet 2005; 67: 451–467.
Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, et al. Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 2004; 41: 892–899.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Ropers HH, Hoeltzenbein M, Kalscheuer V, Yntema H, et al. Nonsyndromic X-linked mental retardation: where are the missing mutations?. Trends Genet 2003; 19: 316–320.
Bauters M, Van Esch H, Marynen P, Froyen G . X chromosome array-CGH for the identification of novel X-linked mental retardation genes. Eur J Med Genet 2005; 48: 263–275.
Lugtenberg D, de Brouwer AP, Kleefstra T, Oudakker AR, et al. Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J Med Genet 2006; 43: 362–370.
Poteet-Smith CE, Smith JA, Lannigan DA, Freed TA, et al. Generation of constitutively active p90 ribosomal S6 kinase in vivo: implications for the mitogen-activated kinase-activated protein kinase family. J Biol Chem 1999; 274: 22135–22138.
Trivier E, De Cesare D, Jacquot S, Pannetier S, et al. Mutations in the kinase Rsk-2 associated with Coffin–Lowry syndrome. Nature 1996; 384: 567–570.
Hanauer A, Young ID . Coffin–Lowry: clinical and molecular features. J Med Genet 2002; 39: 705–713.
Bione S, Tamanini F, Maestrini E, Tribioli C, et al. Transcriptional organization of a 450-Kb region of the human X chromosome, in Xq28. Proc Natl Acad Sci USA 1993; 90: 10977–10981.
D'Adamo P, Menegon A, Lo Nigro Cristiana, Grasso M, et al. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat Genet 1998; 19: 134–139.
Emery AEH . Emery-Dreifuss syndrome. J Med Genet 1989; 26: 637–641.
Yorifuji H, Tadano Y, Tsuchiya Y, Ogawa M, et al. Emerin, deficiency of which causes EDMD, is localizad at the inner nuclear membrana. Neurogenet 1997; 1: 135–140.
Robertson SP . FLNA has diverse roles in embryonic, fetal and postnatal development. Molecular pathology of filamin A: diverse phenotypes, many functions. Clin Dysmorphol 2004; 12: 123–131.
Gerar-Blanluet M, Sheen V, Machinis K, Neal J, et al. Bilateral periventricular heterotopias in an X-linked dominant transmission in a family with a two affected males. Am J Genet 2006; 140: 1041–1046.
Van Esch H, Bauters M, Ignatius J, Jansen M, et al. Duplication of the MECP2 regions is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Genet 2005; 77: 442–453.
Billuart P, Bienvenu T, Ronce N, des Portes V, et al. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 2002; 4: 191–199.
Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, et al. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 2003; 126: 1537–1544.
des Portes V, Boddaert N, Sacco S, Briault S, et al. Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am J Med Genet 2004; 124: 364–371.
Chabrol B, Girard N, N'Guyen K, Gérard A, et al. Delineation of the clinical phenotype associated with OPHN1 mutations based on the clinical and neuropsychological evaluation of three families. Am J Med Genet 2005; 138: 314–317.
Kutsche K, Yntema H, Brandt A, Jantke I, et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet 2000; 26: 247–250.
Kohn M, Steinbach P, Hameister H, Kehrer-Sawatzki H, et al. A comparative expression analysis of four MRX genes regulating intracellular signalling via small GTPases. Eur J Hum Genet 2004; 12: 29–37.
Ramakers GJ . Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 2002;25:191-199. Erratum in Trends Neurosci 2002; 8: 432.
Shaw-Smith C, Redon R, Rickman L, Rio M, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet 2004; 41: 241–248.
Acknowledgements
This work was supported by the Instituto Carlos III (PI041126, PI050776, PI050159).
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Madrigal, I., Rodríguez-Revenga, L., Badenas, C. et al. MLPA as first screening method for the detection of microduplications and microdeletions in patients with X-linked mental retardation. Genet Med 9, 117–122 (2007). https://doi.org/10.1097/GIM.0b013e318031206e
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318031206e
Keywords
This article is cited by
-
X chromosome and suicide
Molecular Psychiatry (2011)
-
Xq26.2-q26.3 microduplication in two brothers with intellectual disabilities: clinical and molecular characterization
Journal of Human Genetics (2010)
-
Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy
European Journal of Human Genetics (2009)
-
Genetik in der Pädiatrie als Interaktion zwischen Klinik und Labor
Monatsschrift Kinderheilkunde (2008)
-
X-chromosome tiling path array detection of copy number variants in patients with chromosome X-linked mental retardation
BMC Genomics (2007)
