
Abstract
Purpose: To investigate the genetic loci/mutations among the Chinese Charcot-Marie-Tooth disease type 1 (CMT1), which accounts for approximately 70% of Charcot-Marie-Tooth; and to study the genetic heterogeneity and mutation frequency.
Methods: CMT1A duplication and mutations at loci of MPZ, Cx32/GJB1, EGR2, and LITAF/SIMPLE were analyzed among 32 clinically diagnosed CMT1 patients of Chinese ancestry.
Results: The CMT1A duplication was detected in 62.5% (20/32) CMT1 patients. This duplication accounts for the major mutation for Chinese CMT1. Among 12 cases that have no CMT1A duplication detected, three point mutations including one (3.1%) in MPZ and two (6.3%) in Cx32 were identified. No mutation was detected in genes PMP22, EGR2 and LITAF among the remaining nine (28.1%) CMT1 patients.
Conclusion: The mutation frequency for the Chinese CMT1 is similar to that seen in the global ethnic population. Molecular testing of the CMT1A duplication, along with the loci of MPZ and Cx32, may detect the majority of Chinese CMT1 patients.
Similar content being viewed by others


Main
Charcot-Marie-Tooth disease type 1 (CMT1) is a peripheral neuropathy characterized by distal muscle weakness and atrophy, reduced nerve conduction velocities (NCV), and demyelination and re-myelination with onion bulb formation on sural nerve biopsy. Genetically, CMT1 is heterogeneous. Many genes have been identified to underlie CMT1.1 A 1.5-Mb tandem duplication in band 17p11.2 which comprises the gene of peripheral myelin protein 22 (PMP22) (MIM# 601097) is the most frequent cause of CMT1.2,3 In patients who have no PMP22 duplication detected, mutations have been found in the following genes: PMP22, myelin protein zero (MPZ/P0) in 1q23 (MIM# 159440), connexin 32 (Cx32/GJB1) in Xq13.1 (MIM# 304040), early growth response 2 (EGR2/Krox-20) in 19q21.1–q22.1 (MIM# 129010), and lipopolysaccharide-induced transcription factor (LITAF/ SIMPLE) in 16p13.3–p12 (MIM# 603795).4–8
Molecular genetic studies of CMT have been performed widely in variant genetic ethnic background populations,9 but relatively limited in the Chinese. For analyzing the distribution of the mutations that result in CMT1 among Chinese CMT patients, we have examined the CMT1A duplication, mutations in genes PMP22, MPZ, CX32, EGR2 and LITAF from 32 unrelated patients of Chinese ancestry who were clinically diagnosed as having CMT1.
MATERIALS AND METHODS
Patient samples
A total of 32 unrelated Chinese patients, who were clinically diagnosed with CMT1 on a basis of electrophysiological criteria, have been studied. A group of 50 healthy controls were recruited after careful clinical and electrophysiological examinations. The ethnic background of patient and control groups is Han majority of Chinese population. Both groups were collected from Beijing area. Informed consent, according to the protocol approved by the Institutional Ethics Committee, was obtained from all participants.
Genotyping of CMT1A duplication
Genomic DNA from venous blood samples was extracted with standard protocol. Genotyping of the CMT1A duplication was carried out using three microsatellite markers (D17S4A, D17S9A, and D17S9B) as previously reported.10
Mutation screening
For cases that have no CMT1A duplication detected, each patient was studied for the presence of published mutations within the coding regions of PMP22, MPZ, GJB1, EGR2 and LITAF genes. Six coding exons of MPZ were individually amplified by the polymerase chain reaction (PCR), using previously reported PCR primers and PCR conditions.11 The coding region of Cx32 was amplified with three sets of primers as described by Nelis et al.12 Four exons of the PMP22 gene were amplified with a protocol previously described.7 The coding region of EGR2 was amplified with nine sets of primers.13 Exons 2 through 4 of the LITAF (SIMPLE) gene were amplified with a set of primers and PCR conditions reported by Street et al.8
The corresponding PCR products except exon 4 of the LITAF gene were analyzed by single strand conformational polymorphism (SSCP) under the following conditions: five microliters of PCR product and 5 μl SSCP loading buffer were combined; each sample was denatured at 95°C for 10 minutes and then cooled on ice immediately. Each product was analyzed under the following electrophoresis conditions: 8–10% acrylamide (3% bisacrylamide); 5% glycerol; 1 μl TBE; 4°C; 1 mA per 10 bp for 10 hours. Silver staining was then applied. PCR fragments that gave abnormal SSCP patterns were sequenced with ABI 3100 automatic sequencer (Applied Biosystems, USA). The PCR products of exon 4 of the LITAF gene were directly sequenced, without SSCP analysis.
RESULTS
Detection of CMT1A duplication
Through genotyping of three microsatellite markers (D17S4A, D17S9A, and D17S9B) that are located within the chromosome 17p11.2–p12 region, CMT1A duplication was analyzed initially. Three distinct bands or two bands with a clear dosage difference10 were interpreted as having CMT1A duplication, which was detected in 20 CMT1 patients. The distribution of mutations among CMT1 patients in this study is summarized in Table 1.
Mutations at the Cx32 and MPZ genes were found in three CMT1 patients
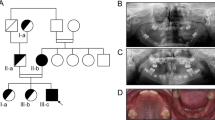
Abnormal SSCP patterns were found in 3 affected patients but not in non-carriers (Fig. 1A–C). DNA sequencing analysis of the PCR product with the abnormal band in Figure 1A showed a G>A exchange at nucleotide 622 in exon 2 of Cx32 resulting in a Glutamate (Glu) to Lysine (Lys) substitution at codon 208 (Glu208Lys) (Fig. 2A). This mutation was detected in a female patient who had family history of an X-linked dominant CMT. The second point mutation corresponding to the abnormal band in Figure 1B was a C>T exchange at nucleotide 643 in exon 2 of Cx32, which resulted in an Arg to Trp substitution at codon 215 (Arg215Trp). This mutation was identified from an isolated male patient (Fig. 2B). Both of the CX32 mutations have been reported previously.14 An intron mutation corresponding to Figure 1C, which caused a splicing error, was detected in the MPZ gene. It was a G>T substitution at -1 of 3′-splice site in intron 3 (c.449-1G>T) (Fig. 2C). This mutation has been reported in a Korean family15 with autosomal dominant classical CMT1 phenotype (Table 2).

SSCP analysis showing the altered mobility pattern of PCR fragments of the Cx32 and MPZ in 3 CMT1 cases. Filled symbols represent subjects with CMT1 and open symbols represent normal subjects. Arrows indicate the abnormal bands. (A) An altered mobility shift corresponding to exon 2 of Cx32 was found in a female patient who had a family history of an X-linked dominant CMT. (B) Altered mobility shifts corresponding to exon 2 of Cx32 were found in an isolated male patient. (C) An altered mobility shift corresponding to exon 4 of MPZ was detected in an isolated female patient.

Causative mutations in Cx32 and MPZ identified by DNA sequencing. Filled symbols represent subjects with CMT1 and open symbols represent normal subjects. Arrow indicates the mutation site. (A) Corresponding to Figure 1A, a G>A exchange at nucleotide 622 in exon 2 of the Cx32 gene resulting in a glutamate to Lysine substitution at codon 208 (Glu208Lys) was detected by sequencing the reverse strand of the PCR products. (B) Corresponding to Figure 1B, a C>T exchange at nucleotide 643 in exon 2 of the Cx32 gene resulting in a Arg to Trp substitution at codon 215 (Arg215Trp) was detected by sequencing the reverse strand of the PCR products. (C) Corresponding to Figure 1C, a G>T exchange at -1 of 3′-splice site in intron 3 (c.449-1G>T) of the MPZ gene was detected by sequencing the forward strand of the PCR products.
No point mutation found among PMP22, EGR2 and LITAF genes
Mutation screening with SSCP among genes PMP22, EGR2 and LITAF was performed in the remaining nine patients who have no mutation detected in either CMT1A duplication or point mutation in genes Cx32 or MPZ. Although we did not find any mutation in PMP22 and EGR2 genes, one variation c.274A>G (Ile92Val) was detected in gene LITAF (data not shown) among four patients who were heterozygous. This variation was reported as a polymorphism previously.16
DISCUSSION
Molecular genetic studies of CMT have been widely performed in different ethnic background populations, but relatively limited in the Chinese. In the present study, we investigated 32 CMT1 Han-Chinese patients.
CMT type 1 (CMT1) accounts for about 70% of CMT and the majority mutation for it is associated with the CMT1A duplication. We detected the CMT1A duplication and found the duplication frequency in Chinese patients was 62.5% (Table 1). This is similar to those (53.6–70.7%) reported in global ethnic CMT1 groups,15,18–23 but is higher than 31.2% in Japanese CMT1 patients.17
Twelve patients without CMT1A duplication were further screened for mutations in genes PMP22, MPZ, Cx32, EGR2 and LITAF. Two mutations (Glu208Lys and Arg215Trp) in Cx32 and one mutation (c.449-1G>T) in MPZ were identified. These three mutations have been reported previously,14,15 but were not reported in the Chinese as yet. Our finding of these three mutations in the Chinese CMT1 patients indicates that there is no ethnic difference regarding the distribution of the mutation.
The mutation frequency in Cx32 (6.3%) and MPZ (3.1%) evaluated in this study was analogous to those (Cx32, 5.7–l7.4%; MPZ, 2.4–4.6%) presented in several European and Asian groups (Table 1),15,18–23 but was lower than that of the Japanese (Cx32, 10.9%; MPZ, 9.4%).17
In our current study, no mutation was detected in coding regions of the PMP22, EGR2 and LITAF genes. Only one variation (Ile92Val) in the LITAF gene, which was not considered as a causative mutation but a polymorphism,16 was detected in 4 of 12 patients in our study. The frequency of this variation is the same as that previously reported.16 Population data showed that the mutation frequencies of the PMP22 (1.2–4.7%) and EGR2 (0.7–3.6%) genes were low15–19,22 when analyzing the mutations in LITAF among 192 American CMT patients. Their results indicated that the mutation in LITAF is not a common cause of CMT.
In addition to the duplication of CMT1A and the mutations in genes PMP22, MPZ, Cx32, EGR2 and LITAF, many other mutations and chromosome loci, such as NEFL, GDAP1, MTMR2, PRX, LMNA related to CMT, have been reported (reviewed by Zhang and Zhong).1 The CMT cases, in which we did not find the underlying genetic defect in our current study, may be caused by the genes mentioned here, although we did screen the LMNA locus,24 for which further investigation should be performed. Nevertheless, finding the CMT1A duplication, along with the mutation at genes MPZ and Cx32, accounts for the majority of Han-Chinese CMT1 patients, as seen in the global ethnic population. This made it possible to offer clinical molecular testing for the Chinese CMT1 patients.
References
Zhang YZ, Zhong N . Molecular genetics of Charcot-Marie-Tooth disease. J Peking University (Health Science) 2005; 37: 100–105.
Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991; 66: 219–232.
Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). Neuromuscul Disord 1991; 1: 93–97.
Bellone E, Di Maria E, Soriani S, Varese A, et al. A novel mutation (D305V) in the early growth response 2 gene is associated with severe Charcot-Marie-Tooth type 1 disease. Hum Mutat 1999; 14: 353–354.
Bergoffen J, Scherer SS, Wang S, Scott MO, et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 1993; 262: 2039–2042.
Hayasaka K, Himoro M, Sato W, Takada G, et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet 1993; 5: 31–34.
Roa BB, Garcia CA, Suter U, Kulpa DA, et al. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med 1993; 329: 96–101.
Street VA, Bennett CL, Goldy JD, Shirk AJ, et al. Chance, Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology 2003; 60: 22–26.
Szigeti K, Garcia CA, Lupski JR . Charcot-Marie-Tooth disease and related hereditary polyneuropathies: Molecular diagnostics determine aspects of medical management. Genet Med 2006; 8: 86–92.
Latour P, Boutrand L, Levy N, Bernard R, et al. Polymorphic short tandem repeats for diagnosis of the Charcot-Marie-Tooth 1A duplication. Clin Chem 2001; 47: 829–837.
Nelis E, Timmerman V, De Jonghe P, Vandenberghe A, et al. Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene. Hum Genet 1994; 94: 653–657.
Nelis E, Simokovic S, Timmerman V, Lofgren A, et al. Mutation analysis of the connexin 32 (Cx32) gene in Charcot-Marie-Tooth neuropathy type 1: identification of five new mutations. Hum Mutat 1997; 9: 47–52.
Van Der Zwaag B, Verzijl HT, Beltran-Valero De Bernabe D, Schuster VL, et al. Mutation analysis in the candidate Mobius syndrome genes PGT and GATA2 on chromosome 3 and EGR2 on chromosome 10. J Med Genet 2002; 39: E30.
Fairweather N, Bell C, Cochrane S, Chelly J, et al. Mutations in the connexin 32 gene in X-linked dominant Charcot-Marie-Tooth disease. Hum Mol Genet 1994; 3: 29–34.
Choi BO, Lee MS, Shin SH, Hwang JH, et al. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2004; 24: 185–186.
Saifi GM, Szigeti K, Wiszniewski W, Shy ME, et al. SIMPLE mutations in Charcot-Marie-Tooth disease and the potential role of its protein product in protein degradation. Hum Mutat 2005; 25: 372–383.
Numakura C, Lin C, Ikegami T, Guldberg P, et al. Molecular analysis in Japanese patients with Charcot-Marie- Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Hum Mutat 2002; 20: 392–398.
Mostacciuolo ML, Righetti E, Zortea M, Bosello V, et al. Chart-Marie-Tooth disease type 1 and related demyelinating neuropathies: Mutation analysis in a large cohort of Italian families. Hum Mutat 2001; 18: 32–41.
Mersiyanova IV, Ismailov SM, Polyakov AV, Dadali EL, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat 2000; 15: 340–347.
Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, et al. Estimation of the mutation frequencies in Charcot- Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet 1996; 4: 25–33.
Bort S, Nelis E, Timmerman V, Sevilla T, et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and heredit7ry neuropathy with liab7lity to pressure palsies. Hum Genet 1997; 99: 746–754.
Boerkoel CF, Takashima H, Garcia CA, Olney RK, et al. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol 2002; 51: 190–201.
Wise CA, Garcia CA, Davis SN, Heju Z, et al. Molecular analyses of unrelated Charcot- Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am J Hum Genet 1993; 53: 853–863.
Song S-J, Zhang Y-Z, Chen B, Wang M-J, et al. No mutation was detected in the LMNA gene among sporadic Charcot-Marie-Tooth patients. J Peking University (Health Science) 2006; 38: 78–79.
Acknowledgements
This work is supported in part by the Chinese Ministry of Sciences and Technology (2004BA720A03), Chinese Ministry of Health 985 Project (985-2-035-39), and the Chinese Ministry of Education 211 Project. We would like to thank all participants for their participation.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Song, S., Zhang, Y., Chen, B. et al. Mutation frequency for Charcot-Marie-Tooth disease type 1 in the Chinese population is similar to that in the global ethnic patients. Genet Med 8, 532–535 (2006). https://doi.org/10.1097/01.gim.0000232481.96287.89
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.gim.0000232481.96287.89
Keywords
This article is cited by
-
Four novel mutations of the myelin protein zero gene presenting as a mild and late-onset polyneuropathy
Journal of Neurology (2010)
-
U1 snRNA mis-binding: a new cause of CMT1B
neurogenetics (2010)
