
Abstract
Disclaimer: ACMG standards and guidelines are designed primarily as an educational resource for physicians and other health care providers to help them provide quality medical genetic services. Adherence to these standards and guidelines does not necessarily ensure a successful medical outcome. These standards and guidelines should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the geneticist should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen. It may be prudent, however, to document in the patient’s record the rationale for any significant deviation from these standards and guidelines.
Similar content being viewed by others

Main
Pompe disease is a rare, progressive, and often fatal muscular disease. The underlying pathology is a deficiency of the enzyme acid alpha-glucosidase (GAA) that hydrolyzes lysosomal glycogen. Pompe disease is a single disease which manifests as a clinical spectrum that varies with respect to age at onset, rate of disease progression, and extent of organ involvement. The advent of enzyme replacement therapy for this condition will necessitate early diagnosis. This guideline for the management of Pompe disease was developed as an educational resource for health care providers to facilitate the prompt and accurate diagnosis and treatment of patients. An international group of experts in various aspects of Pompe disease met to review the evidence base from the scientific literature and their expert opinions. Consensus was developed in each area of diagnosis, treatment and management in the development of this guideline. This management guideline specifically addresses evaluation and diagnosis across multiple organ systems (cardiology, pulmonary, gastrointestinal/nutrition, musculoskeletal, neurological) involved in both infantile and late-onset Pompe disease. Conditions to consider in a differential diagnosis stemming from presenting features and diagnostic algorithms are provided. Aspects of functional assessment, rehabilitation, nutritional management, emerging therapeutics, care coordination, nursing, genetic counseling, prenatal diagnosis and screening also are addressed. A guideline that will facilitate the appropriate diagnosis, treatment and management of patients with Pompe disease was developed. It will raise awareness of this condition and the presentation of patients across the disease spectrum in order to expedite their diagnosis so they can take advantage of emerging therapeutics such as enzyme replacement therapy (ERT).
GENERAL/BACKGROUND
Pompe disease, also referred to as acid maltase deficiency (AMD) or glycogen storage disease type II (GSDII), is an autosomal recessive disorder caused by a deficiency of the lysosomal enzyme acid-α-glucosidase (GAA).1 It was the first recognized lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease. In Pompe disease, lysosomal glycogen accumulates in many tissues with skeletal, cardiac, and smooth muscle most prominently involved.2,3
All patients with Pompe disease share the same general disease course, namely the steady accumulation of glycogen substrate in target tissues leading to progressive debilitation, organ failure and/or death; resulting in a spectrum of disease severity. Severity varies by age of onset, organ involvement including degree and severity of muscular involvement (skeletal, respiratory, cardiac), and rate of progression. Due to the presence of weakness and hypotonia, it has also been classified as a neuromuscular disease or metabolic myopathy. Great phenotypic variability has led to the creation of types based on the age of onset and degree of organ involvement. These subtypes have been referred to inconsistently in the literature by various terminologies such as infantile, late-infantile, childhood, juvenile, and adult-onset form. Herein, we have considered Pompe disease a continuum of disease spectrum varying by age of onset, organ involvement and degree of myopathy. In an attempt towards establishing a uniform terminology, we broadly classify Pompe disease as follows:
infantile form that includes
-
patients as originally described by JC Pompe, a Dutch pathologist, in 19324 who exhibit rapidly progressive disease characterized by prominent cardiomegaly, hepatomegaly, weakness and hypotonia, and death due to cardiorespiratory failure in the first year.5,6 This represents the most severe end of the disease spectrum and is often referred to as classic infantile Pompe disease.
-
patients with the infantile variant form (nonclassic infantile Pompe disease) with slower progression and less severe cardiomyopathy but presenting in the first year of life as described originally by Hers1 and, more recently, by Slonim et al.7 and which has also been variably classified in the literature with the infantile or childhood forms (discussed below).
late-onset form that includes
-
childhood, juvenile, or muscular variant that is a heterogeneous group usually presenting later than infancy and typically not including severe cardiomyopathy.
-
adult-onset form characterized by a slowly progressive myopathy predominantly involving skeletal muscle that can present as late as the second to sixth decade of life.
However, it is important to recognize that age of onset does not always delineate subtypes well; occasional so called “juvenile-onset” or mild variant cases may present prior to 12 months of age. Thus the clinical presentation must be considered along with the age of onset when classifying cases.
Incidence data are limited with reports ranging from 1 in 14,000 to 1 in 300,000 depending upon ethnicity or the geographic area studied.2 The infantile form has an apparent higher incidence among African-Americans and Chinese2 whereas the late-onset adult form has a higher incidence in The Netherlands.8 In the Netherlands, the incidence of the infantile-onset form is 1/183,000. The combined incidence of all forms of Pompe disease is estimated to be 1:40,000.2,8,9
Until recently, there has been no specific treatment for Pompe disease, other than supportive care. Alanine, high-protein diets, branched chain amino acids and β-agonists such as albuterol10–14 have been reported to offer limited benefits in some individuals. Epinephrine and glucagon, which enhance cytosolic glycogen breakdown, have also been attempted with no therapeutic effect.15 Bone marrow transplantation has not been shown to be useful, though experience is very limited.16 Similarly, heart transplant for which there is even less experience has not been shown to be useful (personal communication, James Leonard, 2006).
Natural history
The clinical presentation of Pompe disease ranges from a rapidly progressive infantile form which is uniformly lethal to a more slowly progressive late-onset form. In general, disease severity typically correlates inversely with residual acid α-glucosidase (GAA) activity. Patients with infantile Pompe disease present in the first few months of life with a hypertrophic cardiomyopathy, generalized muscle weakness and hypotonia, followed by death from cardiorespiratory failure usually by 1 year.1,2 Two recent studies delineate the natural history of infantile-onset Pompe disease. The natural history of classic infantile Pompe disease in 20 original Dutch cases and 133 cases from the literature has been reported.17 In this study, the median age at symptom onset was 1.6 months in both groups; the median age at diagnosis (and death) was 5.3 (7.7) and 4.5 (6.0) months in the Dutch cohort and in the literature cases, respectively. In the second study the natural history of disease progression was determined from a retrospective chart review on a worldwide cohort of 168 patients with infantile-onset (classic and non classic infantile) Pompe disease. In this study, the median age at symptom onset was 4 months, first ventilator support occurred at 5.9 months, and death occurred at 8.7 months. Despite intervention, mortality remained largely unchanged over the decades.6,18
In both studies, cardiomegaly, cardiomyopathy, hypotonia, muscle weakness, respiratory distress, respiratory infections, feeding difficulties, and failure to thrive were reported as frequently presenting signs and symptoms.5,6,18 In summary, both studies concluded that the natural history of infantile Pompe disease has changed little since its original description, though the absence of large, well characterized and unbiased data sets has been a limitation.
Late-onset Pompe disease can present at any age and is characterized by a lack of severe (typically absence of) cardiac involvement and a less dismal short-term prognosis.2 Symptoms are related to progressive skeletal muscle dysfunction. Proximal lower limb and paraspinal trunk muscles are usually affected first, followed by involvement of the diaphragm and accessory muscles of respiration. In some cases, diaphragmatic weakness may be evident before any other significant weakness is noted.19 As the muscle weakness worsens, patients often become wheelchair users and may require assisted ventilation. Respiratory failure is usually the cause of significant morbidity and mortality in this form of the disease. The age of death varies from early childhood to late adulthood, depending on rate of disease progression, extent of respiratory muscle involvement and other comorbidities.20
PURPOSE
This guideline is intended as an educational resource. It highlights current practices and therapeutic approaches to the diagnosis and management of the multiple complications of Pompe disease, including emerging therapies such as enzyme replacement therapy and other potential therapies.
METHODS/PROCESS
Consensus development panel
An international group of experts in the a) clinical and laboratory diagnosis, b) treatment and management (cardiac, respiratory, gastrointestinal/dietary, musculoskeletal, neurologic, psychosocial, anesthesiologic, general medical, and supportive and rehabilitative); c) clinical trials of enzyme replacement therapy and d) genetic aspects of Pompe disease was assembled in order to review the evidence base and develop management guidelines. Following a meeting during which published material and personal experience were reviewed by the panel, experts in the various areas reviewed the literature in these areas and drafted their appropriate guideline sections. Conflict of interest statements were provided by the participants. However, it is important to appreciate that many experts may have conflicts when rare disease treatments are tied to orphan drug designations and their related marketplace protections. All members of the panel reviewed and approved the final guidelines. Consensus was defined as agreement among all members of the panel. For the most part, the evidence and resulting recommendations are considered expert opinion since additional levels of evidence were not available in the literature. Where available, evidence from clinical trials is used to guide recommendations. Penultimate drafts of these guidelines were shared with an external review group consisting of Rochelle Hirschhorn, MD, Gregory Carter, MD, Johan van Hove, MD, PhD, James Leonard, MD, Jennifer Li, MD, and Yuan-Tsong Chen, MD, PhD. Their suggestions were considered by the working group and changes were made as considered appropriate by the working group.
Target audience
This guideline is directed at a wide range of providers. Although care is commonly provided by metabolic disease specialists/biochemical geneticists and neuromuscular experts, it is important that primary care providers and other specialists who are often involved in the care of these patients be able to recognize these patients in order to intervene and provide the appropriate care.
DIAGNOSTIC CONFIRMATION
Differential diagnosis
Clinical
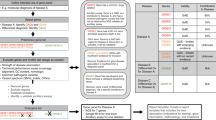
The diagnosis of Pompe disease often poses a diagnostic dilemma due to the rarity of the condition and the relatively nonspecific nature of the phenotypic features that may only in aggregate lead to suspicion of Pompe disease. The diseases that are important in the differential diagnosis of infantile and late onset Pompe disease are shown in Tables 1 and 2 respectively. Diagnostic algorithms that stem from key presenting features for infantile-onset and late onset Pompe disease are shown in Figures 1 and 2, respectively.21 After diagnosis, the interval for clinical follow-up of Pompe disease patients must be individualized for the patient, but a general guide of close follow up for infantile Pompe disease patients and every six months for older and late-onset Pompe disease patients is useful.

Diagnostic algorithm for infantile onset Pompe disease.

Diagnostic algorithm for late onset (> 1 year) Pompe disease.
Clinical evaluation
Infantile and Late-Onset Pompe Disease
A chest x-ray and electrocardiogram (ECG) are valuable screening tests in the diagnostic algorithm for infantile Pompe disease and an echocardiogram is a valuable next step. Chest x-ray shows massive cardiomegaly. ECG shows a short PR interval as well as very tall QRS complexes. In a series of 19 patients with infantile onset Pompe disease a short PR interval was noted in 14 cases (∼75%).22 Caution in interpreting an ECG with attention to voltage calibration is advised, since QRS complex voltage may be very high, and a valuable diagnostic clue may be missed if the operator has turned the gain down. Age appropriate norms and adjustment for gestational age for the PR interval should also be used. Late onset cases rarely display cardiomegaly on a chest x-ray or ECG which minimizes their utility in these patients. In infantile Pompe disease echocardiogram typically reveals a hypertrophic cardiomyopathy with or without left ventricular outflow tract obstruction in the early stages of the disease. In the late stages of infantile disease, patients may have impaired cardiac function and a dilated cardiomyopathy. Among patients presenting with cardiomyopathy, an electromyogram (EMG) can be useful to document presymptomatic myopathy. Blood tests should include a serum creatine kinase (CK) examination. CK elevation is a sensitive although very nonspecific marker for Pompe disease. The greatest elevation is usually found in infantile-onset patients (as high as 2000 UI/L). Approximately 95% of late-onset patients have an elevated CK23; however, some adults with Pompe disease may have CK levels within the normal reference range. Serum enzymes such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), or lactate dehydrogenase (LDH) may be elevated and may reflect enzymes released from muscle.24 There is increasing evidence of the value of a specific glucose tetrasaccharide in urine [Glc4 (Glcα1-6Glcα1-4Glcα1-4Glc)] in the evaluation of Pompe disease.25 Like CK, Glc4 is a sensitive though nonspecific marker for Pompe disease. It can also be elevated in other glycogen storage diseases and clinical correlation is needed. As the clinical presentation in late-onset cases is typically that of a proximal myopathy, much of the evaluation involves laboratory testing of muscle (i.e., EMG, nerve conduction studies and/or muscle biopsy for histology, histochemistry and enzyme assay). Many of the laboratory tests that are useful in the evaluation of suspected infantile onset Pompe disease are also valuable in considering the late onset form (i.e., CK, AST, ALT, LDH and Glc4). Pulmonary function testing can identify respiratory compromise in both.
Laboratory testing
Diagnostic Testing
The clinical diagnosis is traditionally confirmed by the virtual absence (infantile-onset) or markedly reduced (late-onset) GAA activity in tissues such as cultured fibroblasts from skin biopsy, muscle biopsy, purified lymphocytes, mononuclear cells and lymphoid cell lines. However, historically GAA enzyme measurement is most reliably performed in cultured fibroblasts or muscle, due to the possibility of alternate isoenzyme activities (e.g., maltase-glucoamylase as discussed below) masking disease in white cell assays. Acid alpha glucosidase (GAA) has an optimum activity at pH 3.7 to 4.5 and hydrolyzes alpha 1-4 and alpha 1-6 bonds in glucose polymers. In a typical assay, GAA activity is measured at an acidic pH (3.7 to 4.5) and compared with the activity of neutral glucosidase at pH 7.0, using maltose and glycogen or the maltose fluorescent synthetic analogue, 4-methylumbelliferyl-α-D-glucoside (4-MUG) as substrates.
Measurement of GAA activity in skin fibroblasts is the current gold standard. Cultured fibroblasts are obtained from a skin biopsy which is then grown to confluency prior to the enzyme assay. This can take up to 4–6 weeks, and can thus significantly delay the diagnosis. Muscle tissue biopsy, although more invasive, allows muscle acid-α-glucosidase activity and glycogen content to be assayed directly and rapidly. It can also be used for histological studies to assess the location and amount of glycogen accumulation in the tissue. Great care in handling is required so as not to wash out glycogen and alter cell and tissue pathology. In late-onset Pompe disease, the site of muscle biopsy can impact results due to the variability of glycogen accumulation both between different muscles and between the muscle fiber type within a muscle. Furthermore, there is anesthesia risk (see section below) in infantile Pompe disease if an open biopsy is done.26 This risk should be carefully considered, particularly if the patient is already shown to have severe cardiomyopathy and skeletal myopathy.
Muscle biopsy in Pompe disease shows the presence of vacuoles that stain positively for glycogen. In advanced stages of the disease, glycogen accumulation is seen both in the lysosomes and dispersed in the cytosol. Quantitatively, muscle glycogen content is elevated up to tenfold above normal in infantile Pompe disease and to a lesser extent in late onset patients.
In the past, leukocytes isolated from whole blood have not been a reliable tissue for the measure of GAA activity owing to interfering maltases.27 The most significant interference has been recognized as the maltase-glucoamylase (MGA) activity found in polymorphonuclear leukocytes. An improved assay has been described that uses an isolated lymphocyte population and acarbose as an inhibitor of MGA (personal communication, Rhona Jack, PhD), though it may require further validation.
New methods have been developed that assay GAA activity in dried blood spot (DBS) extracts. Isolation of GAA from DBS extracts by immunocapture28 or competitive inhibition of MGA activity using maltose29,30,32 or acarbose31,32 have been used to remove the interfering MGA activity. DBS can be conveniently collected by the heel- or finger-stick method and shipped from locations remote from the analytical center.32 These novel DBS methods are not only suitable for newborn screening of Pompe disease, but also offer a rapid, noninvasive first tier diagnostic test for ruling out Pompe disease. Blood can be directly spotted onto filter paper and, in situations where access to filters is not available, whole blood can be sent to a diagnostic laboratory. Such tests in clinical investigation are best accessed by specialists who are aware of their limitations and they may save valuable time in patient care and treatment.
Elevation of Glc4 in urine has been identified in patients with Pompe disease. Diagnostically, it has close to 100% sensitivity in identifying patients with infantile Pompe disease.25,33 As a biomarker, it has been shown to correlate well with the clinical response to ERT.33,34 Analysis of urinary Glc4 is recommended as a noninvasive ancillary diagnostic test for Pompe disease. This method has also been validated for dried urine on filter paper, which facilitates collection and shipment from locations remote from an analytical center. When combined with the aforementioned GAA enzyme assay in DBS, the diagnostic sensitivity should be close to 100% for infantile Pompe disease. It is equally important to note that negative results from both tests would almost certainly rule out a diagnosis of infantile Pompe disease and eliminate the need for more invasive or time-consuming procedures such as skin or muscle biopsies. These assays are done in specialized laboratories only and are currently not widely available outside of clinical investigational studies.
In general, GAA enzyme activities of <1% of normal controls are seen in the infantile form of Pompe disease. In the late onset forms, activities from 2 to 40% of normal controls are seen and can vary across tissues with muscle tissue showing wider ranges due to the variability in protein content in muscle as compared to fibroblasts. Clinical correlation and molecular diagnosis are often needed however, as there are reports of late onset patients with <1% GAA activity in skin fibroblasts. Laboratory testing to confirm a diagnosis of Pompe disease thus includes several blood tests, some of which may preclude the need for more invasive testing. A diagnostic algorithm including testing to consider the infantile form is presented in Figure 1 and for the late onset form in Figure 2.
Mutation Analysis: While enzyme activity analysis remains the diagnostic test of choice for individuals with Pompe disease, mutation testing has important uses. It is particularly useful in the identification of carriers when a familial mutation is known. There are also some common mutations with useful genotype-phenotype correlations, though many are less specific or are of such infrequent occurrence as to be less useful in this context.35–38 Due to the potential overlap of residual GAA enzyme activity in late-onset Pompe with heterozygotes, molecular analysis of the GAA gene may be required to confirm the diagnosis. This is increasingly important with the advent of ERT and other therapies in development for Pompe disease. More than 100 mutations and numerous variants in the GAA gene have been identified and are catalogued by the Erasmus University Medical Center in Rotterdam, Netherlands (http://www.eur.nl/FGG/CH1/Pompe/). The mutations are spread across the gene though mis-sense mutations in exon 14 may be over represented. Among the recurrent mutations in the infantile-onset cases is a single base pair deletion, Δ525T, that is seen in 9% of U.S. cases.37 This same mutation accounts for 34% of Dutch cases.38 The exon 18 deletion mutation is seen in infantile-onset cases and accounts for about 25% of Dutch and Canadian cases but only about 5% of U.S. cases.37,38 The leaky IVS1(-13T->G) splice-site mutation accounts for about 50% of late-onset cases. There are some populations in which particular mutations are more common due to founder effects while allelic heterogeneity can be significant in admixed populations as exist in the U.S. The R854X mutation is found in many African American and African cases; D645E is seen in many Chinese infantile cases; the 2741AG->CAGG insertion is seen in Turkish cases; and the G925A mutation is seen in many European cases.
OVERVIEW OF MANAGEMENT
Pompe disease is a multi-system disorder and is best managed by a multidisciplinary team led by a physician with experience managing this disorder. Team members should include a metabolic disease specialist/biochemical geneticist in addition to the specialists dictated by the disease manifestations, which might include a cardiologist, pulmonologist, neurologist, neuromuscular specialist, intensivist, orthopedist, respiratory therapist, physical therapist, occupational therapist, otolaryngologist, speech therapist, audiologist, genetic counselor, and a metabolic dietitian. All specialists involved in the care of an individual with Pompe should have an understanding of the disease, its broad and protean manifestations, and challenges, including the psychological and emotional impact of this devastating disease on patients and families. There should be a team member with experience in Pompe disease (e.g., the metabolic specialist) who is able to coordinate the patient’s care.
CARDIOLOGY
The common cardiac issues occurring in Pompe disease include cardiomyopathy, heart failure and arrhythmia. There is also a component of cardiac dysfunction which contributes to respiratory failure from a hemodynamic standpoint and from the impact of cardiac size on lung capacity.
Cardiomyopathy
In infantile Pompe disease, the effects of glycogen accumulation are very pronounced in the heart. Lysosomal glycogen accumulation results in a significant amount of cardiac hypertrophy (between +2 and +10 or higher z-score for LV mass) that may begin in utero and that is significant even at 4–8 weeks of age (personal communication, Carolyn Spencer, MD; Jami Levine, MD; Barry Byrne, MD, PhD, 2005). The cardiac response to glycogen accumulation can result in hypertrophic or hypertrophic and dilated cardiomyopathy. Beyond infancy there is variable involvement of the heart in Pompe disease and a spectrum of disease among children with partial enzyme activity ranging from unaffected to moderate cardiac hypertrophy and cardiac dysfunction (personal communication, Carolyn Spencer, MD; Jami Levine, MD; Barry Byrne, MD PhD, 2005). In patients with late onset Pompe disease there is generally no clinically identifiable heart disease. However, there are no published data of systolic and diastolic function in this group of patients. There are case reports of late-onset Pompe patients with cardiac disease which was thought to be influenced by the glycogen storage39 yet this is the exception rather than the rule.
Electrophysiological findings and potential for arrhythmia
Interference with specialized conducting tissues produces a shortening of the atrio-ventricular (P-R) interval on the ECG.39 The mechanism of accelerated atrioventricular conduction in Pompe disease may be related to the insulator effect of glycogen in conduction tissue40,41 however glycogen accumulation can also result in a short PR in association with other models of glycogen storage.42 True Wolf-Parkinson-White syndrome has been reported to occur in Pompe disease43 as well, though a coincidental occurrence can not be ruled out. These conduction abnormalities, in conjunction with the hypertrophic cardiomyopathy, place these patients at high risk of tachyarrhythmia and sudden death, especially in situations of stress such as infection, fever, dehydration and anesthesia.26,40 Twenty-four hour ambulatory ECG (Holter monitoring) is thus useful in the management of Pompe disease patients.44
Routine evaluation in Pompe
The assessment of cardiac function by echocardiography is an essential part of the ongoing management of Pompe patients. Initial findings of increased wall thickness and left venticular (LV) mass need to be evaluated on a regular basis by a standardized approach since establishing the stage of the cardiomyopathy is useful in determining treatment. This also can help with following disease progression and response to intervention. The LV mass evaluated by two dimensional measurements using the length-area relationship has provided the most reliable assessment. The absolute mass value (not indexed for BSA) is compared to a normal control population to yield a z-score which is an indicator of how far the patient exceeds the mean left ventricular mass in normal subjects. Functional measures including 2-D ejection fraction (EF) and the myocardial performance index are helpful in longitudinal follow-up.
Management
Cardiomyopathy in patients with Pompe disease should be treated cautiously and preferably by a pediatric cardiologist who has experience with the disease. Care needs to be individualized and based on the stage of disease, since inappropriate use of the standard drugs used to treat cardiomyopathy may worsen an already compromised heart. Infants who present with Pompe disease in the earlier phases of the disease generally present with severe ventricular hypertrophy with or without left ventricular outflow tract obstruction and normal or even hyperdynamic left ventricular EF. In the presence of LV outflow tract obstruction, the use of digoxin, other inotropes, diuretics, and afterload reducing agents such as ACE-inhibitors may exacerbate the left ventricular outflow tract obstruction. These agents, however, are generally used in the later phases of the disease in the face of ventricular dysfunction.3 Several anecdotal unreported cases of sudden death associated with the use of beta blockers in Pompe patients have suggested that this class of drugs should be used judiciously. Furthermore, in any case of cardiac hypertrophy there is potential for arrhythmia which may be due to underlying subendocardial ischemia from inadequate coronary perfusion of a massively hypertrophied ventricle. Therefore, special care must be taken in Pompe disease patients to avoid hypotension in diagnostic procedures requiring anesthesia (see below) or when there is volume depletion.26 Reduction in cardiac mass is observed during ERT and reverse remodeling does occur to varying degrees leading to changes in cardiac function as measured by EF. A transient fall in EF has been noted in the first 12–24 weeks of therapy and therefore measurement of myocardial performance index is useful. Development of ventricular ectopy on ERT has been noted, underscoring the need for monitoring with twenty-four hour ambulatory ECG’s at regular intervals.44
PULMONARY
As Pompe disease progresses, the muscles weaken leading to low lung volumes, an impaired cough, blood gas abnormalities, and sleep-disordered breathing. These changes are similar to those seen in any patient with an underlying neuromuscular weakness, such as Duchenne muscular dystrophy and spinal muscle atrophy.45 Patients with Pompe disease are also at an increased risk for aspiration pneumonia (see GI section below). While disease progression in patients with the infantile form is rapid,5,6,17 patients with late onset Pompe disease have a more gradual onset of similar, but initially less severe, respiratory compromise.
Progressive loss of pulmonary function
Approximately 60% of patients with late-onset Pompe disease have a mild reduction in vital capacity (<80% predicted) and 30–40% have moderate reduction (<60% predicted).35,46,47 Although the pattern of muscle weakness is variable, a rough correlation has been observed between measures of pulmonary function and proximal muscle weakness, particularly in the lower extremities.47 Diaphragmatic involvement can be an early and prominent finding in Pompe disease and respiratory failure often develops while patients are still ambulatory; it may even be the initial clinical manifestation of the disease.35,46,48–50 Diaphragm weakness may be characterized by a decrease in the vital capacity (VC) while the patient is in the supine versus prone position (a 10% decrease in VC indicates mild diaphragmatic weakness; a 30% decrease indicating severe weakness).50 Diaphragmatic dysfunction and VC are predictive of the development of chronic sleep hypoventilation and respiratory failure. Objective assessment of pulmonary function in infants is technically difficult, thus complete pulmonary evaluation requires assessment of clinical factors such as daytime energy-level and degree of fatigability (i.e., the ability to feed without developing increased work of breathing).
Impaired cough due to abdominal muscle weakness
Impaired cough results in retained secretions and an inability to clear both the normal volume of pulmonary secretions as well as those associated with acute infections, thereby predisposing the patient to developing atelectasis and pneumonia. Patients with infantile onset disease are especially vulnerable to left lower lobe atelectasis secondary to the enlarged heart compressing the left main stem bronchus. In patients with late onset Pompe disease measurement of the maximal expiratory pressure (MEP), maximal inspiratory pressure (MIP), expiratory reserve volume, and peak cough flow provide objective measurements of muscle strength while for those with early onset disease, measurement of the Negative Inspiratory Forced Maneuver (NIFM) or MIP may be used to obtain this information.
Progressive respiratory insufficiency
Chronic CO2 retention (elevated arterial pCO2 and serum bicarbonate concentration) and hypoxemia (decreased arterial pO2 and oxygen saturation) are seen as muscle weakness progresses, usually developing first while asleep, and then later when awake. In late onset patients, respiratory insufficiency may only be minimally perceived, especially when exercise capacity is limited secondary to skeletal muscle weakness.
Sleep disordered breathing
This is often present in patients with Pompe disease with respiratory muscle weakness because of the mechanical disadvantage of the supine position and the effect of sleep on respiratory control mechanisms. Decrease in upper airway tone, especially during REM sleep, may further compromise ventilation. Nocturnal hypoventilation commonly precedes daytime respiratory failure. In patients with late onset disease, this may occur while the upright vital capacity is only moderately abnormal due to the disproportionate diaphragmatic involvement.47 Pompe patients may also have an impaired ability to compensate for any obstructive sleep apnea that may be present51–53 leading to worsened hypercapnia and hypoxemia.41,54 Progression to cor pulmonale and cardiorespiratory failure is possible. In infantile Pompe disease, sleep disordered breathing may develop before the parents report symptoms.55
Routine evaluation of respiratory function
Assessment of the patient’s respiratory status should be done at each visit, with emphasis on the patient’s cough, the presence of wheezing or shortness of breath, exercise tolerance, energy level, degree of fatigueability, and in infants, their ability to feed. Physical examination should focus on the respiratory rate, the use of accessory muscles of respiration, adequacy of air exchange, presence or absence of adventitial breath sounds, and quality of the patient’s cough. In late onset cases, pulmonary function should include spirometric measurement of VC, forced expiratory volume in 1 second (FEV1), MIP and MEP. Diaphragm weakness can be further evaluated by measurement of the supine vital capacity. For infantile onset disease, spirometry is not an option and infant pulmonary function testing may require sedation, which is not advisable. When available, measurement of crying vital capacity (CVC) can give reproducible measurements of the infant’s vital capacity. Negative inspiratory flow measurement (NIFM), a parameter similar to the MIP, gives a measurement of inspiratory muscle strength.56 Gas exchange should be evaluated in all patients to assess for the development of respiratory failure. Pulse oximetry and capnography should routinely be performed. If normal, arterial blood gas analysis is usually not necessary. If capnography is not available, a venous bicarbonate and/or pCO2 or capillary blood pCO2 should be obtained to assess for alveolar hypoventilation. Chest radiographs may be useful for determining the presence of atelectasis and help determine where chest physical therapy needs to be directed. Radiographs should be obtained upon diagnosis and if there is clinical deterioration (a significant change in auscultation or worsening of O2 saturation and/or ventilation parameters).
Routine evaluation of sleep respiratory function
All patients with Pompe disease should have a detailed sleep history obtained at diagnosis and at follow up appointments. The American Academy of Sleep Medicine recommends full polysomnography for all sleep evaluations.57 Alternative studies (such as pulse oximetry and/or capnography) are insufficient to assess for sleep disordered breathing, though they may be used to assess for nocturnal hypoxia or hypoventilation, respectively.57 Oximetry alone is not a sufficient screening modality for the assessment of sleep disordered breathing.57–59 Symptoms suggesting sleep disordered breathing include snoring, apneas, gasping respirations, restless sleep, irritability, or excessive daytime somnolence (the latter in late onset patients). Detailed examination of the nasopharynx and oropharynx should assess for the presence of (possible) obstruction. Patients should be objectively assessed for obstructive sleep apnea or hypoventilation at diagnosis with polysomnography, regardless of symptoms history. Treatment of respiratory insufficiency and obstructive sleep apnea should be followed with some type of monitoring so as to document its effectiveness. Follow up sleep studies should be performed when clinically indicated.57
Management
Addressing impaired secretion clearance due to weakened cough is critical to maintaining optimal functional status. Both manual and mechanical techniques used in treating other neuromuscular disorders should be used to assist cough and preserve pulmonary function.45,55 Liberal use of bronchodilators should be used in conjunction with these airway clearance techniques and assisted coughing maneuvers (i.e., The Cough Assist In-Exsufflator)60 to maximize the patient’s pulmonary toilet care. Inspiratory muscle training may also be important.45 Bronchodilators and steroids (both inhaled and/or oral) can be useful when an asthmatic process is present. If there is underlying cardiomyopathy, the use of selective beta agonists (e.g., levalbuterol) may be recommended. All pulmonary infections should be aggressively treated.
Supplemental O2 (either nocturnal or continuous) can be used to treat hypoxia, provided that hypoventilation is not present. If hypoxia occurs only during sleep, management is controversial. While most practitioners give nocturnal O2 for the hypoxia, it seems reasonable that patients with obstructive sleep apnea be first managed with support of the upper airway (nasal C-PAP), and that those with nocturnal hypoventilation be managed with nocturnal noninvasive positive pressure ventilation (Bi-level ventilation) before O2 is added. This approach has been shown to be effective and well tolerated in infants with Pompe disease.55 If hypoventilation is present while both awake and asleep, consideration should be given to using continuous noninvasive positive pressure ventilation or even tracheostomy tube placement with mechanical ventilation.
GASTROINTESTINAL/NUTRITION
Patients with Pompe disease have feeding and swallowing difficulties and often fail to thrive. In infantile Pompe disease, contributing factors include facial hypotonia, macroglossia and/or tongue weakness, poor oral range of motion with extremely limited tongue elevation and decreased ability to achieve tongue cupping and lip seal for sucking. These patients have difficulty with saliva management and are less vigilant with regard to swallowing causing pooling of secretions and drooling. Vocal quality is often noted to be “wet” indicating pooling of secretions at the level of the vocal cords, correlating with increasing respiratory complications which could be secondary to aspiration events. In patients with late onset Pompe disease, fatigue of jaw muscles with difficulty swallowing a bolus and chewing food is often a first complaint and this often results in an inadequate intake of total calories, vitamins, and minerals leading to endogenous muscle protein breakdown.
Swallowing dysfunction can be diagnosed in patients with Pompe disease by a videofluoroscopic swallowing assessment which identifies impairments in the different phases of swallowing and should be done at baseline in all newly diagnosed cases. In the oral phase of the swallow study, deficits include decreased sucking strength, oral dysmotility, and/or delayed initiation of the swallow reflex to the level of the valleculae or pyriform sinus. Mild to moderate residue after completion of the swallow is noted in the pharyngeal phase. A 4 point scale can be used to assess the degree of dysfunction in the oral and pharyngeal phase of swallow.61
Patients with Pompe disease are at increased risk for aspiration. By using a scale such as the Penetration/Aspiration Scale the risk for laryngeal penetration and/or presence of aspiration can be assessed.62 Penetration of liquid into the laryngeal vestibule and aspiration has been documented as early as age 4 months in babies with Pompe disease (personal communication, Priya Kishnani, MD 2006). Management by a feeding team that includes a speech pathologist and a dietician (preferably one experienced in metabolic diseases) is of paramount importance in developing and monitoring a feeding plan. If problems are noted in the swallow study, modification of dietary texture with commercial thickeners or cereals may be utilized to provide a safe feeding regimen. If, however, the risk for aspiration is high, oral feeds may need to be discontinued and the patient gavage fed. Evaluation for gastroesophageal (GE) reflux should be undertaken when indicated to determine site of gavage feeding. In instances when there is significant GE reflux, nasoduodenal or gastro-jejunal feeding is recommended rather than nasogastric feeding to minimize the risk of aspiration. One should avoid long surgical procedures, such as a Nissen fundoplication because of high anesthesia risk and low success rate in patients with an underlying myopathy. In some instances a combination of oral and tube feeding to allow for adequate intake of calories, and to allow for normal oral sensory development may be recommended.
Oral stimulation and non-nutritive sucking for nonoral feeders should be provided to maintain normal oral sensory development and to develop emerging oral skills. Modified dietary textures should be re-evaluated on a regular basis to maintain safety in light of a changing disease process. Improvements in swallowing dysfunction have been noted in some infantile-onset patients receiving ERT and patients have been able to resume oral feeding. Growth parameters such as height, weight and head circumference should be followed closely. Changing the texture and on occasion pureeing food has also been found to be extremely useful in the late onset cases.
For patients with late-onset Pompe disease, the goal is intended to manage 1) the increasing accumulation of glycogen and 2) the increase in amino acid utilization.7,11 Slonim et al.63,64 have observed that a high protein-low carbohydrate diet plus aerobic exercise, can be beneficial to some of these patients (see cautions on limits of exercise in the Musculoskeletal/Functional/Rehabilitation section). The rationale to this form of therapy is an attempt to decrease glycogen deposition, increase muscle fatty acid utilization, and at the same time compensate for the increased amino acid oxidation that has been shown to occur in Pompe disease.63 A study by Bodamer,13 however, failed to demonstrate improvement in their late-onset Pompe disease patients treated with a high protein diet and alanine supplementation. A high protein diet may be a good adjunct to ERT but randomized controlled studies are needed. Overall, maintaining good nutrition with attention to macro and micronutrients is important in the management of all patients with Pompe disease.
MUSCULOSKELETAL/FUNCTIONAL/REHABILITATION
Musculoskeletal system and function
Musculoskeletal involvement in Pompe disease is characterized by progressive weakness that leads to decreased motor function, altered postural tendencies and positioning, and the use of compensatory patterns of movement. Secondary musculoskeletal impairments such as contracture, deformity, and osteoporosis can occur and further compromise function. Gross and fine motor function may be limited by weakness, decreased endurance, secondary musculoskeletal impairment, and cardiorespiratory impairment (see Case & Kishnani article in this issue for details).65 Communication and interaction may be compromised by oral-motor weakness that impairs speech, articulation, and phonation and by decreased respiratory function for support of phonation and sound production.
Pathology of muscle weakness
Myopathy results from glycogen accumulation in muscle, leading to compromised muscle function and progressive weakness. Muscles may feel firm or hypertrophic despite weakness.2,3 Distribution and extent of weakness vary depending on severity of disease with weakness usually greater proximally than distally and greater in lower extremities than in upper extremities. Weakness is usually symmetrical but imbalanced across joints. Weakness that affects pulmonary and respiratory function is covered in the pulmonary/respiratory section. Weakness that affects oral motor function in feeding is covered in the gastrointestinal/nutrition section.
Pathokinesiology of muscle function and secondary musculoskeletal impairment
Profound generalized weakness in infantile onset Pompe disease can limit all movement or most antigravity movement. Imbalanced weakness leads to alterations in posture and movement used to mechanically lock joints and compensate for weakness.66 Momentum is used to maximize kinematic advantage. Although compensations are effective in maximizing function initially, persistent use of compensation over time can lead to biomechanical disadvantage, contracture, and deformity leading to increasing weakness and disability.67,68 With severe, early weakness, the unopposed influence of gravity has the most profound effect on positioning and the development of contracture and deformity. With mild to moderate weakness, imbalanced weakness and compensatory movement patterns and postures have an increased role in the development of contracture and deformity.
Osteopenia and osteoporosis
Osteopenia and osteoporosis have recently been recognized as complications of Pompe disease. Thoracic vertebral fracture was reported in the case study of an adult patient with late onset disease who had osteopenia, hypomagnesemia, hypocalcemia, and hypocalciuria.69 Femoral and thoracic vertebral fractures have been identified in patients with infantile onset Pompe disease.70 Osteopenia has been seen in patients with Pompe disease as young as 4 months of age, which could be attributed to chronic immobilization and weakness, but osteopenia has also been identified in patients with Pompe disease with good motor strength and nutrition71 and needs to be further studied. The pathophysiologic mechanisms of osteopenia/osteoporosis in Pompe disease are not yet well understood so are managed generically.71,72
Routine evaluation in Pompe (see Appendix A):
Multidisciplinary evaluation should include assessment within each category of the World Health Organization (WHO) International Classification of Functioning, Disability, and Health (ICF).73 Specific assessments are outlined in Appendix A.
Rehabilitation management of Pompe disease
Rehabilitation management of Pompe disease should be comprehensive and preventative, based on an understanding of the pathokinesiology of disease progression and on individual assessment.74 It should optimize and preserve motor and physiological function, prevent or minimize secondary complications, promote and maintain the maximum level of function, and maximize the benefits of ERT and other therapies when they become available. Rehabilitation services should include physical therapy, occupational therapy, speech therapy, respiratory therapy, orthopedics, early intervention/educational/vocational rehabilitation and independent living services, and the use of adaptive equipment, assistive technology, and orthotic intervention where indicated. Rehabilitation related to respiratory and pulmonary issues are discussed within those sections of this guideline. Management of feeding as relates to oral motor function is addressed in the gastrointestinal/nutrition section.
Strengthening
Guidelines for muscle strengthening are not established for individuals with Pompe disease. Studies of the effect of strengthening in individuals with Pompe have been few, with small numbers and recommend the use of submaximal and aerobic exercise.63,64,75 Caution regarding exercise in the presence of myopathy and cardiorespiratory compromise is important. Excessively strenuous exercise is believed to be detrimental in most diseases characterized by muscle deterioration, as it may increase degeneration, and should be avoided. In Pompe, additional potential, theoretical concern exists that excessive contraction could increase leakage of glycogen from lysosomes or cause lysosomal rupture.76
Medical stability should be established prior to initiation of therapeutic activity, with appropriate subsequent monitoring of cardiopulmonary response to activity level, exercise, and position (especially supine). Precautions regarding strengthening of fragile muscles should be followed using guidelines from other degenerative muscle diseases,77,78 including an emphasis on the use of submaximal and aerobic exercise; avoidance of excessive resistive and eccentric exercise; use of functional activities for exercise; and avoidance of overwork weakness, disuse atrophy, and excessive fatigue. Although excessively strenuous exercise should be avoided, gentle submaximal exercise is believed to be important to allow practice of new motor skills, especially in children; to allow gentle strengthening within physiological limits; to avoid additional disuse atrophy, especially in muscles not be used spontaneously because of biomechanical compromise and relative weakness compared to other muscles; and to avoid secondary cardiopulmonary compromise and deconditioning from inactivity. In addition, there is evidence that submaximal exercise may stimulate the degradation of some of the glycogen that accumulates in the cytosol.75 Intervention for enhancing muscle function should also include strategies to optimize biomechanical advantage and use of energy conservation techniques. If sufficient clearance of glycogen occurs with ERT, a more complete capacity for exercise may emerge. There is evidence in the Pompe mouse model that ERT may differentially clear glycogen from cardiac and type I muscle fibers more efficiently and more fully than from type II fibers,79 and this finding may have an impact on strengthening.
Exercise should be performed with appropriate monitoring, with rests established as needed. Facilitation of functional movement with graded assistance may allow practice of appropriate developmental skills and strengthening within physiological limits in infants. For individuals with greater strength, gentle aerobic and submaximal functional exercise is generally considered beneficial.
Prevention of contracture and deformity
Principles for treatment of contracture and deformity in neuromuscular disorders are well established80 and should be followed for individuals with Pompe disease. Contracture and deformity should be prevented by counteracting deforming forces with the use of gentle forces over time including daily stretching, correction of positioning, use of splinting and orthotic intervention, provision of adequate support in all positions, especially sitting and supported standing as appropriate, and education of patients and families.80 Adaptive equipment and orthoses can be essential in the control of contracture and deformity and can provide changes in position and pressure relief for maintenance of skin integrity in individuals who cannot shift their own weight or change positions independently. Orthotic intervention often includes the use of ankle foot orthoses (AFO’s) to prevent plantar flexion contractures,81 thigh binders to prevent iliotibial band contractures, knee splints to prevent knee flexion contractures, and resting wrist/hand/finger splints to prevent hypoextensibility in long wrist/finger flexors over multiple joints and flexor contracture at the wrist or in individual finger joints. Seating systems in adapted strollers or wheelchairs are critical to prevent or minimize contracture and deformity, especially spinal deformity, and should include a solid seat and back, hip guides, lateral trunk supports, knee adductors, and head support as needed, and as individually assessed. Supported standing is considered beneficial to prevent or minimize lower extremity contracture and osteoporosis82 and may include the use of supine, prone, vertical and hydraulic standers or the use of power standing capabilities on motorized wheelchairs. Power tilt, recline and elevating leg rests on motorized wheelchairs can allow independent position change and weight shift for assistance in minimizing contracture and maintaining skin integrity. Orthopedic surgery may need to be considered in some cases (such as scoliosis management), in which progression occurs beyond that which is amenable to conservative management. Anesthesia precautions should be followed during orthopedic surgery.
The use of positioning, splinting, and standing devices should be coordinated with medical specialists due to potential contraindications such as cardiac or pulmonary compromise, osteoporosis, the risk of fracture, hip subluxation or dislocation, and prohibitive contractures. Consideration must be given to the number of hours per day that a muscle is in a shortened position, as this will determine the risk for the development of contracture and deformity and guide successful prevention.83
Osteopenia monitoring and management
Emerging reports of osteopenia, osteoporosis, and fracture, suggest that screening patients with Pompe disease for osteopenia is indicated. Screening should include a DEXA (dual energy x-ray absorptiometry). Care in interpreting the results of DEXA in infants and children, is needed to ensure that the results are compared with age- and gender–matched controls. At the current time there is insufficient evidence to suggest the (z scores) frequency at which DEXA scans should be obtained in patients with Pompe disease. The frequency should be individualized although DEXA should not be performed more frequently than every 6 months. Factors that have the potential to contribute to osteopenia and osteoporosis have therapeutic implications. Nutrition needs to be adequate especially in terms of intake of calcium and Vitamin D and attention given to medications (e.g., long term use of certain diuretics which could cause hypercalciuria, long term steroid use). Potential interventions may include the provision of weightbearing in physical therapy and in standing devices.71,72 There is insufficient evidence to suggest pharmacologic therapy such as bisphosphonates as a preventive treatment in Pompe disease at the current time. However, it may be reasonable to consider introducing these agents in children having Pompe disease and osteoporosis once the pathophysiology is better understood and more of these medications have been proven to be safe and effective in children with other causes of osteoporosis.
Oral motor and respiratory management related to communication
Intervention may be needed for the development of speech, language, and communication in infants, children and adults with oral motor and respiratory involvement who are too weak to readily initiate, practice, and use vocal sound production for communication, to maximize speech and communication for individuals who are on ventilators or have tracheostomies, and for those demonstrating speech delay. Positioning and support to maximize functional use of the respiratory system for sound production should be done. Augmentative communication, including signing and the use of assistive technology, such as voice output systems, may be needed for function, to compensate for weakness and decreased endurance, and to allow conservation of energy. Availability and use of computers (including voice activated systems) and internet access can facilitate communication.
Function
At every age and stage, appropriate function and maximal levels of independence, governed by medical stability, should be encouraged. Functional developmental activities and independence in the activities of daily living should be supported. Family education is essential in fostering appropriate independence. The use of mobility devices such as canes, walkers, manual wheelchairs, and powered mobility are usually added gradually in late onset disease, often used part-time at first, for long distance community mobility or on uneven terrain. Motorized mobility should be considered early in infantile onset Pompe disease, at developmentally appropriate ages.84 Adapted car seats can be used for infants and children, and those designed for infants with poor head control should be used for safe transport of fragile infants. Appropriate assistive technology should be provided with training in its use at home and in educational and work tasks in order to maximize function, functional independence, safety, access and participation. Driving may be appropriate, depending on age and abilities.
Provision of service
Rehabilitation services may be provided in the home, in center-based programs, at school, in inpatient facilities, and in outpatient settings, depending on the needs and preferences of the individual and the family. Services, adaptive equipment, and assistive technology may be funded by numerous different agencies, as outlined below under care coordination.
NEUROLOGICAL
Central nervous system
The natural history of Pompe disease largely reflects the pathologic consequences of glycogen accumulation in skeletal and cardiac muscle, but autopsy studies have also demonstrated glycogen accumulation in the brain, brainstem nuclei and anterior horn cells.2 Cognitive function is usually thought to be normal in both infantile and late onset disease, but it is possible that subtle impairments might become apparent if the natural history of infantile Pompe disease is preferentially modified by emerging therapies such as ERT.
Anterior horn cell involvement is manifest as EMG signs of denervation in these children.2 Clearance of muscle glycogen by ERT leading to improved skeletal muscle function and prolonged survival might theoretically lead to the emergence of late onset Pompe/spinal muscular atrophy phenotypes in such cases and a long-term follow up of these patients is needed. Affected individuals should be followed using appropriate standardized instruments as described above. Motor unit number evaluation (MUNE) has shown promise in studies of SMA, and should be considered as an adjunctive assessment of anterior horn cell function in future studies of patients receiving ERT. (see appendix 1 for discussion of quantitative muscle testing, electromyography, histopathology, use of the Pompe Pediatric Evaluation of Disability Inventory (PEDI), and functional motor testing (timed tests, arm and leg functional testing)).
Hearing loss has been recognized in some infantile Pompe disease patients, and may reflect direct involvement of the cochlea (rare), the conductive apparatus (common), or both.85 In a series of 20 patients with infantile Pompe disease seen at Duke University, nearly 80% of the ears tested demonstrated Distortion Product Otoacoustic Emissions (DPOAE) suggestive of normal or near normal cochlear function. 73% of the ears tested had abnormal tympanometry consistent with middle ear dysfunction. None of the patients in this series had absent DPOAE’s in the presence of normal tympanometry, decreasing the likelihood of cochlear involvement (personal communication, Priya Kishnani, MD, and Gwen O’Grady, MA, 2006). In addition to standard neonatal hearing screening, children should have baseline, routine, and annual hearing tests. Testing should include behavioral assessment when possible, otoacoustic emissions, tympanometry, and Auditory Evoked Potentials (ABR/BAER) using air and bone conducted stimuli. Individual rather than normative data should be used to determine site of lesion of the hearing loss due to the extreme variability seen in the normative data available in the pediatric (<18 months) population and the maturational effects observed in evoked potential testing.86 Careful interpretation of tests by audiologists with pediatric experience is necessary as children with infantile Pompe disease are difficult to accurately assess due to their motor and other delays as well as the increased incidence of middle ear involvement. As with all pediatric patients, clinical circumstances such as frequent episodes of otitis media (OM) may dictate more frequent examinations. Aggressive medical therapy of OM and chronic serous OM should be pursued, with surgical intervention reserved for treatment failures (note: see cautions in “Surgery and Anesthesia”). Hearing re-evaluation should be performed following medical/surgical intervention to ensure remediation of any potential conductive component. Hearing aids should be used when indicated because permanent hearing loss has been identified.
Some patients with Pompe disease experience fever of uncertain origin, presumed to be of central origin. As outlined in “General Medical Care” all patients should be thoroughly evaluated for sources of infection and considered for empiric treatment until a negative workup for infectious and inflammatory disorders is completed. There are also reports of glycogen accumulation in vascular smooth muscle resulting in aneurysms/rupture of blood vessels such as the basilar artery87,88 internal carotid artery and medial cerebral arteries in patients with late-onset Pompe disease.89,90 This has resulted in focal weakness and in the extreme instance even death. Vascular involvement of intracranial blood vessels should be considered in the work up and evaluation of patients with Pompe disease, especially those that are symptomatic.
GENERAL MEDICAL CARE
Due to the overall hypotonia, cardiomyopathy (in infantile presentation) and respiratory muscle weakness, individuals with Pompe disease are at very high risk for pneumonia and other infections which may lead to respiratory failure, intubation with ventilator dependence and even death. Infections need to be managed very aggressively in this population and the threshold to treat infections should be low. Immunizations should be kept up to date and patients and household members should receive the influenza vaccine during influenza season. Furthermore, palivizumab (Synagis) should be used during respiratory syncytial virus (RSV) season, in infants and ‘young children with Pompe disease as this can be life-threatening in these patients. Pneumococcal vaccine (Prevnar) is currently part of the routine immunization schedule, however the 23 valent peumococcal vaccine should be administered after 2 years of age and for older patients who have not previously received it. Strict hand washing precautions should be implemented to avoid infections. Medical attention should be sought for common symptoms such as a cough or fever as it may indicate a more serious underlying condition. Another important consideration is careful use of over the counter medications to treat colds, cough and other URI symptoms as they often contain sympathomimetic agents which can be detrimental to the heart. The risk benefit ratio of use of medications such as steroids (risk of progressive muscle weakness and osteopenia) and loop diuretics (ototoxicity and calciuria) must be considered prior to administration.
Similarly, infectious agent precautions and aggressive management of infections should be the rule in late-onset patients.
SURGERY/ANESTHESIA
Providing safe anesthesia for infants with Pompe disease is a challenge due to the severe hypertrophic cardiomyopathy and the resultant hemodynamic alterations. Myocardial ischemia and decreased cardiac output are not uncommon events during anesthetic management in infantile Pompe disease. The thickened ventricular wall results in a higher left ventricular end-diastolic pressure (LVEDP) at lower ventricular volumes and an increased potential for subendocardial ischemia. Maintaining a normal coronary perfusion pressure is thus of paramount importance.26 Intraoperative cardiac arrest during halothane anesthesia and sevoflurane inhalation induction followed by a maintenance infusion of propofol in infantile Pompe has previously been reported.91,92 With the advent of emerging therapies such as ERT, survival of these patients has increased and more surgical procedures are being performed. The patients need intensive monitoring and thus surgical procedures should be done with extreme caution and conducted at sites with experienced pediatric/cardiac anesthesiologists. Avoidance of intubation is generally preferred but may be unavoidable in certain circumstances (e.g., macroglossia). When multiple procedures are needed, they may be done as a group, when appropriate, under a single anesthesia. Careful attention to peri-operative fluids is necessary as the dynamic left ventricular outflow tract (LVOT) obstruction can be exacerbated in the presence of hypovolemia. However, overly aggressive fluid resuscitation may lead to pulmonary edema in patients with elevated LVEDP. Echo-derived indices of 2DLV mass and ventricular cavity volume, in addition to EF, should be used as parameters of myocardial function in planning an anesthetic, as EF alone may be misleading in this patient population. Five lead ECG with continuous ST segment monitoring and placement of an indwelling arterial catheter should be strongly considered, if cardiac disease is advanced. The goals of anesthesia are to maintain a higher filling pressure for adequate preload and a normal to high systemic vascular resistance to ensure effective coronary perfusion. Anesthetic agents should thus be chosen to address the marked myocardial hypertrophy and its physiological effects on preload and afterload. Ketamine maintains systemic vascular resistance (SVR) and contractility and is less likely to reduce preload. It is an example of an agent which offers a safer profile in this patient population.92 Etomidate is another acceptable induction agent.93 Inhalational agents and thiopental, should be used with caution and be reserved for patients with less myocardial hypertrophy. Propofol, particularly in higher doses administered to achieve general anesthesia, rather than sedation, may not be the ideal anesthetic agent for Pompe patients, since afterload reduction and a lower diastolic pressure may predispose these infants to the risk of myocardial ischemia. Deaths have been reported (unpublished data) with propofol as the anesthetic agent. Because of the underlying muscle weakness Pompe patients may be more sensitive to neuromuscular blockade. Agents such as suxamethonium should be avoided in Pompe disease as in all myopathic patients due to the potential risk of rhabdomyolysis and hyperkalemia. Malignant hyperthermia precautions should be taken. After recovery from anesthesia infants may be managed on the ward or in an intensive care unit depending on clinical status.
CARE COORDINATION AND NURSING
Care coordination of individuals with Pompe disease is essential to the support and treatment of the patients and their families and may be dictated by the patient’s local demographics and individual needs. Local customs, insurance requirements and other factors contribute to decisions of who assumes this role. Nurses, genetic counselors, local primary care providers, medical geneticists and metabolic disease specialists and neurologists may take a central role in providing culturally sensitive care.94,95 Social workers may also facilitate and coordinate some of the services. Individuals assuming the various roles should have current knowledge of the disease process as discussed in previous sections, as well as a comprehensive understanding of the patient’s needs in the therapeutic settings and the community. Education of team members is often required to teach the family and other resource personnel about the disease process and needed medical procedures, and the anticipated future needs and cautions for optimum care of the patient.
Recognizing that the “family” is the constant in a patient’s life, the plan of care should be centered on needs identified by the family and medical/therapeutic providers working as a team. Parent/family/patient collaboration with the professional team should be facilitated at all levels of care to allow for equal participation in establishing the goals of care. Multiple needs which may include separation from core family members due to hospitalization or therapy, transportation, lodging costs, loss of work days, specialized equipment including an adapted vehicle, household assistance, regular medical procedures, vocational retraining, and educational programming should be addressed. Information about therapies, treatments, and resources should be continuously shared with the family in an appropriate and supportive manner that is sensitive to family relationships and respectful of the patient’s position in the family. Timely referrals to appropriate community agencies, including a social worker or other case worker, and advocating for the needed community services is essential support that can be facilitated by the individual coordinating the care of the patient.
The coordinator, in consultation with the physician providing care should recognize the specialized medical and therapeutic care that is required by the patient and should be able to triage and refer them to specific providers as needed. The strengths, preferences, and coping mechanisms of the patient and family should be respected and incorporated into the needs assessment. In addition, any cultural, religious, language or racial differences and sensitivities should be recognized in the development of the plan of care.
Parent to parent and patient to patient support has been reported by many to be one of the most valued resources provided. Resources for this support are outlined in the following section. Another consideration is to identify support for siblings, grandparents and other family members.
The care coordinator should insure that the patient/family is aware of the medical support that has been coordinated, provide the family with the appropriate contact information, and with the patient/ family permission, facilitate appropriate information sharing. The family and all providers should be updated on the care plan and patient progress at regular intervals. This will insure comprehensive coordinated care that is family centered.
Family needs and resources
The added responsibilities of living with Pompe disease extend beyond the patient often to a vast network of family and friends. Pompe patients want not simply to live, but to thrive- and many of them do just that. To achieve this, there are constant strains and challenges which take many forms - financial, educational, emotional and psychological. There is today a vast network of resources and information, both public and private, that can ease the burdens of this disease and add greatly to the Pompe patient’s quality of life.
The Muscular Dystrophy Association
The Muscular Dystrophy Association (MDA) is a not-for-profit organization whose goal is to fight neuromuscular diseases through a nationwide research effort, a program of patient medical services and extended education for professionals and families. Pompe disease is one of about 40 neuromuscular diseases included within the scope of the MDA’s mission and services. The MDA maintains 235 hospital-affiliated clinics with nurses, physicians and therapists experienced in neuromuscular diseases. Pompe patients may access all of these services.
In addition, for children, the MDA offers special programs that directly offset the cost of medical equipment, including wheelchairs and other mobility assistive devices. The MDA also sponsors over 90 summer camps which are free of cost across the United States for children with neuromuscular disease. These camps offer children with all diseases, including Pompe, the chance to play with other special needs kids in a fun and safe environment.
The MDA also has extensive knowledge and resources related to the challenges of families living with neuromuscular diseases, including Duchenne muscular dystrophy, Pompe disease and spinal muscular atrophy (SMA). Pompe families will find of particular benefit a book published by MDA entitled “Learning to Live with Neuromuscular Disease—a message for parents.”96; This book, published first in 1998, addresses such topics as: taking on the challenge; facing your feelings; the importance of reaching out; strength in family relations; and, what your child needs. The book can be found on the MDA web site (see below).
Indeed, the challenges that most Pompe patients and their families face are enormous. They often times strain relationships, friendships and marriages – sometimes they even break them. This stress is a natural part of the shared burdens that surround the care of a person with Pompe. Those families that deal best with these life-altering challenges are enriched by the understanding and mutual concern that becomes a compelling and dominant force in their lives.
The MDA web site contains useful information. See www.mdausa.org/publications/learning/ or the MDA can be contacted by telephone at 1-800-572-1717 (US) or 520-529-2000 (international).
Other agencies and web sites specific to related diagnoses such as DMD and SMA can provide additional information including: Parent Project Muscular Dystrophy (http://www.parentprojectmd.org), Families of SMA (http://www.fsma.org), and the SMA foundation (www.smafoundation.org).
Medicaid
Medicaid is a joint state-federal entitlement program which provides primarily medical care to low-income Americans. Medicaid has evolved over time and now offers many benefits for providing home and community services to patients with long-term care needs, such as many of those living with Pompe. Medicaid gives each state broad discretion to cover virtually all long-term care services that people with disabilities need to live independently in both home and community settings.
Eligibility for Medicaid varies by state but in every case it is a means-tested entitlement program. For those who qualify, it provides a means for access to a wide range of medical services.
For children with Pompe requiring home nursing and who do not otherwise qualify for Medicaid, other funds may be available. The “Katie Beckett” (or “TERFA”) option is a Medicaid waiver program (http://www.cms.hhs.gov/medicaid/waivers/) that enables states to provide Medicaid specifically for home health care expenses for disabled children and may also cover therapy services, adapted equipment, aide services, and respite care. This is an optional program for each state. Since each state sets its own rules for coverage, please visit www.cms.hhs.gov/medicaid to review each individual state’s scope of coverage and programs. Early Intervention Services are supported by the Early Infant Program for Infants and Toddlers with Disabilities, Part C of the federal Individuals with Disabilities Act (IDEA) which coordinate and fund services for children from birth to 3 years of age (http://www.cec.sped.org/law_res/doc/law/regulations/indexPartC.php). Other federal legislation such as the Public Law 94-142 and the Rehabilitation Act, including Section 504, insure a free public education in the least restrictive environment possible, including the provision of therapy services (PT, OT, ST) when educationally relevant, and assuring access (http://www.ed.gov/policy/speced/leg/edpicks.jhtml?src=ln).
Other valuable government sources of information include the National Information Center for Children and Youth with Disabilities (NICHCY) and the Council for Exceptional Children. NICHCY is a national information and referral resource for families with disability-related issues (www.nichcy.org). The Council for Exceptional Children (www.cec.sped.org) is an international professional organization dedicated to improving educational outcomes for individuals with exceptionalities. Also of value to Pompe families, particularly adult Pompe patients, may be the website www.disabilityinfo.gov. This website offers a significant collection of resources including information on housing, employment, education, income support, technology, and community support.
Other private not-for-profit organizations that provide additional potential sources of support and funding for Pompe patients include The Disabled Children’s Relief Fund (www.dcrf.com). The DCRF focuses on helping children who do not have adequate health insurance, especially the physically challenged. It provides disabled children throughout the United States with assistive devices, rehabilitative services, and support for arts and humanities projects.
Pompe patient organizations
The International Pompe Association (IPA) is a federation of Pompe disease patient groups worldwide. It seeks to coordinate activities and to share experience and knowledge between different groups. Membership in the IPA is open to Pompe and related disease groups. As such, the IPA acts as an umbrella organization for patient organizations globally. One of the most valuable forums for Pompe patients annually is the IPA patient conference held in the Fall of each year.
In the United States, there are two primary Pompe patient support groups: the Acid Maltase Deficiency Association (AMDA) and the United Pompe Foundation. Both groups aim to broaden public awareness and knowledge of Pompe as well as to support select research initiatives at Universities. (www.amda-pompe.org and www.unitedpompe.org).
GENETIC COUNSELING, PRENATAL DIAGNOSIS AND SCREENING
Genetic counseling and prenatal diagnosis
Genetic counseling should be offered to all parents with an affected child with Pompe disease and to all adults with Pompe disease. In counseling families with Pompe disease, at least a three generation pedigree from the consultand or proband should be obtained. Pompe disease is an autosomal recessive condition. De novo mutation rates are expected to be low and parents of an affected individual are assumed to be carriers. The recurrence risk to parents who have had an affected child is 25%. DNA mutation analysis is necessary for the identification of additional family members in the extended family who may be carriers. A small number of laboratories offer DNA diagnostic and/or prenatal diagnostic testing for Pompe disease (see www.genetests.org and http://biochemgen.ucsd.edu/). Some genotype:phenotype correlations have been observed.2,97 Some laboratories may only screen for common mutations rather than provide full gene sequencing. Identification of carrier status in the general population (e.g., in the spouse of a known carrier) is limited and currently not routinely offered. Such individuals can be tested for the common mutations to further refine their risk for having a child with Pompe disease.
Prenatal diagnostic testing is typically performed by measuring enzyme activity in uncultured chorionic villus samples.98,99 Enzyme analysis in amniocytes has also been successfully performed but enzyme activity is typically lower than in chronionic villus studies. Prenatal diagnostic testing by mutation analysis is feasible, it allows for confirmation of the biochemical assay and the absence of contamination of chorionic villus samples, and offers advantages over biochemical methods, if the proband’s mutation has been identified. Therefore, identification of the proband’s GAA mutations, if a sample is available for testing, should be a priority. In equivocal cases, a combination of methods can be used, and some laboratories prefer to offer prenatal enzyme analysis with results confirmed by DNA mutation analysis. When the mutations segregating in the family are known, molecular testing is the gold standard.100 Preimplantation genetic diagnosis (PGD) is also potentially an option for Pompe disease families if specific mutations have been identified.
Screening
The development of Chinese hamster ovary (CHO) cell-derived recombinant human acid alpha glucosidase (rhGAA) 100enzyme replacement therapy that is currently in clinical trials shows promise. Results from several different trials with CHO enzyme and also transgenic enzyme from rabbit milk in earlier trials have shown that patients treated early in the disease, prior to the development of extensive tissue damage, are likely to have the best outcome.101–105 A clinical trial of 18 patients with classic infantile Pompe disease treated with CHO cell derived rhGAA at age ≤ 6 months was recently completed.106 At the end of 1 year of therapy all patients were alive and 83% were alive and free of invasive ventilation. Data from this and other trials were submitted to the Food and Drug Administration for a Biologics License Application in July, 2005. The results from this study build on previous experience and underscore the need for early recognition of affected patients to allow for timely intervention and the best chance for a good outcome. The recent expansion of newborn screening in the United States has led to the early, presymptomatic diagnosis of over 30 different inborn errors of metabolism. The first pilot study of newborn screening for lysosomal storage disorders (Krabbe disease) has begun in New York (though Pompe disease is not in the initial group being tested) and in Taiwan (Pompe disease is included).
Methods for high throughput screening of GAA activity, using the newborn screening dried blood filter specimen (DBS), have been developed and are being validated in large population screening programs.29–31,107 If successful, newborn screening for Pompe disease can become a reality.
ENZYME REPLACEMENT AND OTHER EMERGING THERAPIES
Several open-label clinical trials involving patients with infantile-onset Pompe disease have shown that enzyme replacement therapy significantly prolongs survival,102–110 decreases cardiomegaly, and improves cardiac and skeletal muscle function. In the vast majority of cases cardiac response appears to be good, irrespective of the stage of the disease at the start of ERT. Skeletal muscle response has been more variable than cardiac muscle response. The best skeletal muscle response has been noted in patients treated early, prior to severe skeletal muscle damage. There are several patients with infantile-onset Pompe disease on ERT who are walking, a milestone that would not have been achieved without ERT. Thus with the advent of ERT, the natural history of this once lethal disease has changed and additional physical and mental disabilities may be uncovered. However, there are patients who have not had a good outcome despite early treatment. Factors such as muscle fiber type, stage of disease at start of therapy, genotype and immune response to the recombinant enzyme may play a role in determining outcome and need further investigation. Additional larger-scale trials in infants to establish the extent of long-term benefit, the optimal dosing protocol, and the effect of other factors on outcome of therapy are needed and are currently underway. Trials of ERT in late onset Pompe disease are also underway. With the introduction of therapies such as ERT, long term survival is now a reality and closer attention to the natural history of treated patients is of paramount importance. However, the possibility that neurological manifestations may be uncovered during long-term ERT for infantile onset Pompe disease remains to be resolved.
Progress continues to be made in the development of gene therapy for Pompe disease. Several proof-of-concept studies in Pompe mice demonstrate that various gene delivery vector systems including retrovirus,111 adenovirus112–115 and most recently recombinant adeno-associated viral (AAV) vectors116–121 have potential. Studies demonstrate that expression of the acid alpha glucosidase gene within muscle cells or in some cases secretion of GAA from liver leads to a reduction of the glycogen storage abnormality in the mice. Moreover, these studies showed that the immune response to the expressed enzyme in AAV-treated Pompe mice treated can be reduced in some cases117,121 or be influenced by tissue specific expression.118,119
The use of small molecules to treat lysosomal storage disorders is also emerging as a therapeutic option either as a single agent or in combination with other therapies. Work on a second generation recombinant enzyme for Pompe disease for more efficient targeting to muscle is currently in the preclinical stages. Pharmacologic chaperones that bind to the affected proteins and restore their shape, proper trafficking, and biological activity may also become available for those who can make protein. Several challenges remain before clinical testing of these therapeutic strategies can be considered. In the case of gene therapy, work still remains, especially in terms of safety.
At the current time with the advent of enzyme replacement therapy and the likelihood of its availability in the near future early recognition and appropriate medical management of Pompe disease has assumed new importance. Thus, physicians need to be alert to the signs and symptoms of this disease to facilitate early pick up and allow for therapeutic interventions as they become available. A multidisciplinary team approach should be followed to ensure the best care and outcome for these patients.
In order to optimize recommended disease support measures, a mechanism to collect longitudinal patient data as a means to follow the general disease population and assess the impact of disease support measures as used by physicians around the world is needed. The gold standard for such studies may be the U.S. National Cancer Cooperative Groups that allowed for participation of 85–90% of all children with leukemia and cancer in the U.S. in protocol driven studies with peer reviewed laboratory and clinical data from entry into the studies through outcome. This system was particularly important in providing the statistical power necessary for informing studies and clinical trials for rare diseases. In the absence of such study systems, registries can be useful. The Pompe Registry was launched in September 2004 as a global, observational database to track de-identified natural history and outcomes data for monitoring and analysis by the global Pompe disease medical community. Registry participation is open to all physicians managing patients with a confirmed diagnosis of Pompe disease. The Pompe Registry will act as a repository for disease natural history and select clinical trial data and as a platform to gather prospective data on current and future Pompe patients for a number of years. Information on the registry can be found at www.pomperegistry.com.
References
Hers HG . Alpha-Glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease). Biochem J 1963; 86: 11–16.
Hirschhorn R, Reuser AJJ . Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Beaudet A, Scriver C, Sly W et al. eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill, 2001: 3389–3420.
Kishnani PS, Howell RR . Pompe disease in infants and children. J Pediatr 2004; 144: S35–S43.
Pompe JC . Over idiopatische hypertrophie van het hart. Ned Tijdshr Geneeskd 1932; 76: 304.
van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003; 112: 332–340.
Kishnani P, Hwu W, Mandel H, Nicolino M, et al. On behalf of the Infantile-onset Pompe Disease Natural History Study Group a retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Peds. In press, 2006.
Slonim AE, Bulone L, Ritz S, Goldberg T, et al. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr 2000; 137: 283–285.
Ausems MG, Verbiest J, Hermans MP, Kroos MA, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counseling. Eur J Hum Genet 1999; 7: 713–716.
Martiniuk F, Chen A, Mack A, Arvanitopoulos E, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998; 79: 69–72.
Angelini C, Pegorora E, Marsala SZ, Vergani L, et al. Adult acid maltase deficiency: an open trial with albuterol and branched chain amino acids. Basic Appl Myol 2004; 14: 71–78.
Slonim AE, Coleman RA, McElligot MA, Najjar J, et al. Improvement of muscle function in acid maltase deficiency by high-protein therapy. Neurology 1983; 33: 34–38.
Bodamer OA, Leonard JV, Halliday D . Dietary treatment in late-onset acid maltase deficiency. Eur J Pediatr 1997; 156: S39–S42.
Bodamer OA, Halliday D, Leonard JV . The effects of l-alanine supplementation in late-onset glycogen storage disease type II. Neurology 2000; 55: 710–712.
Bodamer OA, Haas D, Hermans MM, Reuser AJ, et al. L-alanine supplementation in late infantile glycogen storage disease type II. Pediatr Neurol 2002; 27: 145–146.
Hug G, Schubert WK, Soukup S . Treatment related observations in solid tissues, fibroblast cultured and amniotic fluid cells of type II glycogenosis, Hurler disease and metachromatic leukodystrophy. Birth Defects Orig Art Ser 1973; 9: 160–183.
Watson JG, Gardner-Medwin D, Goldfinch ME, Pearson AD . Bone marrow transplantation for glycogen storage disease type II (Pompe’s disease). N Engl J Med 1986; 314: 385.
Kishnani PS, Hwu P, Mandel H, Nicolino M, et al. Onbehalf of the Infantile Pompe natural history group. A retrospective, multinational, multicenter study of the natural history of Infantile Pompe disease. J Pediatr 2006, in press.
Marsden D . Infantile onset Pompe disease: a report of physician narratives from an epidemiologic study. Genet Med 2005; 7: 147–150.
Engel AG, Gomez MR, Seybold ME, Lambert EH . The spectrum and diagnosis of acid maltase deficiency. Neurology 1973; 23: 95–106.
Hagemans ML, Janssens AC, Winkel LP, Sieradzan KA, et al. Late-onset Pompe disease primarily affects quality of life in physical health domains. Neurology 2004; 63: 1688–1692.
Howell RR, Byrne B, Darras B, Kishnani P, et al. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 2006; 8: 289–296.
Ansong A, Li JS, Nozik-Grayck E, Ing R, et al. Electrocardiographic response to enzyme replacement therapy for pompe disease. Genet Med 2006; 8: 297–301.
Ausems MG, Lochman P, van Diggelen OP, Ploos van Amstel HK, et al. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 1999; 52: 851–853.
DiFiore MT, Manfredi R, Marri L, Zucchini A, et al. Elevation of transaminases as an early sign of late-onset glycogenosis type II. Eur J Pediatr 1993; 152: 784.
An Y, Young SP, Hillman SL, van Hove JL, et al. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem 2000; 287: 136–143.
Ing RJ, Cook DR, Bengur RA, Williams EA, et al. Anaesthetic management of infants with glycogen storage disease type II: a physiological approach. Paediatr Anaesth 2004; 14: 514–519.
Dreyfus J, Poenaru L . White blood cells and the diagnosis of alpha-glucosidase deficiency. Pediatr Res 1980; 14: 342–344.
Umapathysivam K, Hopwood JJ, Meikle PJ . Determination of acid-glucosidase activity in blood spots as a diagnostic test for Pompe disease. Clin Chem 2001; 47: 1378–1383.
Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C . Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta 2004; 347: 97–102.
Niizawa G, Blanco M, Casentini C, Borrajo G, et al. Newborn screening for MPS1 and Pompe disease: a pre-pilot study. Mol Genet Metab 2005; 84: A202.
Li Y, Scott CR, Chamoles NA, Ghavami A, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 2004; 50: 1785–1796.
Zhang H, Kallwass H, Young S, Carr C, et al. A comparison of methods for lysosomal acid α-glucosidase activity determination from dried blood spots for diagnosis of Pompe disease. Genet Med 2006; 8: 302–306.
Young SP, Stevens RD, An Y, Chen YT, et al. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution-electrosprayionization tandem mass spectrometry. Anal Biochem 2003; 316: 175–180.
An Y, Young SP, Kishnani PS, Millington DS, et al. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab 2005; 85: 247–254.
Raben N, Plotz P, Byrne BJ . Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002; 2: 145–166.
Laforet P, Nicolino M, Eymard PB, Puech JP, et al. Juvenile and adult-onset acid maltase deficiency in France: Genotype-phenotype correlation. Neurology 2000; 55: 1122–1128.
Hirschhorn R, Huie ML . Frequency of mutations for glycogen storage disease type II in different populations: The del525T and delta exon 18 mutations are not generally “common” in White populations. J Med Genet 1999; 36: 85–86.
Van der Kraan M, Kroos MA, Joosse M, Bijvoet AG, et al. Deletion of exon 18 is a frequent mutation in glycogen storage disease type II. Biochem Biophys Res Commun 1994; 203: 1535–1541.
Wiegand V, Rumpf K, Bardosi A, Meinck H, et al. Hemodynamic findings in the adult form of type II glycogenosis. Z Kardiol 1986; 75: 44–46.
Gillette PC, Nihill MR, Singer DB . Electrophysiological mechanism of the short PR interval in Pompe disease. Am J Dis Child 1974; 128: 622–626.
Metzl JD, Elias ER, Berul CI . An interesting case of infant sudden death: severe hypertrophic cardiomyopathy in Pompe’s disease. Pacing Clin Electrophysiol 1999; 22: 821–822.
Arad M, Moskowitz IP, Patel VV, Ahmad F, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003; 107: 2850–2856.
Bulkley BH, Hutchins GM . Pompe’s disease presenting as hypertrophic myocardiopathy with Wolff-Parkinson-White syndrome. Am Heart J 1978; 96: 246–252.
Cooke A . Ambulatory electrocardiogram analysis in infants treated with recombinant human acid alpha glucosidase enzyme replacement therapy for Pompe disease. Genet Med. In press.
Finder JD, Birnkrant D, Carl J, Farber HJ, et al. American Thoracic Society. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med 2004; 170: 456–465.
Mellies U, Ragette R, Schwake C, Baethmann M, et al. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology 2001; 57: 1290–1295.
Berger KI, Skrinar A, Norman RG, et al. Ventilatory dysfunction in late onset Pompe disease. Proc Am Thorac Soc 2005; 2: A788.
Moufarrej NA, Bertorini TE . Respiratory insufficiency in adult-type acid maltase deficiency. South Med J 1993; 86: 560–567.
Kim DG, Jung K, Lee MK, Hyun IG, et al. A case of juvenile form Pompe’s disease manifested as chronic alveolar hypoventilation. J Korean Med Sci 1993; 8: 221–224.
Keunen RW, Lambregts PC, Op de Coul AA, Joosten EM . Respiratory failure as initial symptom of acid maltase deficiency. J Neurol Neurosurg Psychiatry 1984; 47: 549–552.
American Thoracic Society/European Respiratory Society. ATS/ERS Statement on respiratory muscle testing. Am J Respir Crit Care Med 2002; 166: 518–624.
Redline S, Kapur VK, Sanders MH, Quan SF, et al. Effects of varying approaches for identifying respiratory disturbances on sleep apnea assessment. Am J Respir Crit Care Med 2000; 161: 369–374.
Young T, Palta M, Dempsey J, Skatrud J, et al. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993; 328: 1230–1235.
Margolis ML, Howlett P, Goldberg R, Eftychiadis A, et al. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest 1994; 105: 947–949.
Kravitz RM, Mackey J, DeArmey S, Kishnani PS . Polysomnogram findings in patients with infantile Pompe disease. Proc Am Thorac Soc 2005; 2: A459.
Kravitz RM, Mackey J, DeArmey S, Kishnani PS . Pulmonary function findings in patients with infantile Pompe disease. Proc Am Thorac Soc 2005; 2: A186.
Kushida CA, Littner MR, Morgenthaler T, Alessi CA, et al. Practice parameters for the indications for polysomnography and related procedures: An update for 2005. Sleep 2005; 28: 499–521.
Brouillette RT, Morielli A, Leimanis A, Waters KA, et al. Nocturnal pulse oximetry as an abbreviated testing modality for pediatric obstructive sleep apnea. Pediatrics 2000; 105: 405–412.
Kirk VG, Bohn SG, Flemons W, Remmers JE . Comparison of home oximetry monitoring with laboratory polysomnography in children. Chest 2003; 124: 1702–1708.
Chatwin M, Ross E, Hart N, Nickol AH, et al. Cough augmentation with mechanical insufflation/exsufflation in patients with neuromuscular weakness. Eur Respir J 2003; 21: 502–508.
Rosenbek JC, Robbins JA, Roecker EB, Coyle JL, et al. A penetration-aspiration scale. Dysphagia 1996; 11: 93–98.
Horner J, Riski JE, Ovelmen-Levitt J, Nashold BS Jr . Swallowing in torticollis before and after rhizotomy. Dysphagia 1992; 7: 117–25.
Slonim AE, Rosenthal H, O’Connor MR, Goldberg T, et al. High protein and exercise therapy (HPET) for childhood acid maltase deficiency (AMD). J of Neurological Sciences 1990; 98( Suppl.): A465.
Slonim AE, Bulone L, Minikes J, Hays AP, et al. Benign course of glycogen storage disease type IIb in two brothers. Nature or Nurture? Muscle Nerve 2005; Nov 30 [epublication ahead of print].
Case LE, Kishnani PS . Physical therapy management of Pompe disease. Genet Med 2006; 8: 318–327.
Fowler WM . Rehabilitation management of muscular dystrophy and related disorders: II. Comprehensive care. Arch Phys Med Rehabil 1982; 63: 322–328.
Roy L, Gibson DA . Pseudohypertrophic muscular dystrophy and its surgical management: review of 30 patients. Can J Surg 1970; 13: 13–21.
Archibald KC, Vignos PJ Jr . A study of contractures in muscular dystrophy. Arch of Phys Med and Rehab 1959; 40: 150–157.
Oktenli C . Renal magnesium wasting, hypomagnesemic hypocalcemia, hypocalciuria and osteopenia in a patient with glycogenosis type II. Am J Nephrol 2000; 20: 412–417.
Krishnamurthy VV, DeArmey S, Mackey J, Frush D, et al. Osteopenia in infantile Pompe disease: an unrecognized long term complication. Molecular Genetics and Metabolism 2005; 84: A227.
Quinlivan R, Roper H, Davie M, Shaw NJ, et al. Report of a Muscular Dystrophy Campaign funded workshop in Birmingham, UK, January 16, 2004. Osteoporosis in Duchenne muscular dystrophy; its prevalence, treatment and prevention. Neuromuscul Disord 2005; 15: 72–79.
Biggar WD, Bachrach LK, Henderson RC, Kalkwarf H, et al. Bone health in Duchenne muscular dystrophy: a workshop report from the meeting in Cincinnati, Ohio. July 8, 2004. Neurocuscul Disord 2005; 15: 80–85.
ICF (International Classification of Functioning, Disability and Health) Geneva: World Health Organization (WHO) Press, 2001. http://www.who.int/classifications/icf/en/ (February).
Vignos PJ Jr . Physical models of rehabilitation in neuromuscular disease. Muscle Nerve 1983; 6: 323–338.
Bembi B, Ciana G, Martini C, Bennettoni A, et al. Efficacy of multidisciplinary approach in the treatment of two cases of nonclassical infantile glycogenosis type II. J Inherit Metab Dis 2003; 26: 675–681.
Griffin JL . Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Arch B Cell Pathol Incl Mol Pathol 1984; 45: 23–36.
Eagle M . Report on the muscular dystrophy campaign workshop: exercise in neuromuscular diseases Newcastle, January 2002. Neuromuscul Disord 2002; 12: 975–983.
Fowler WM . Role of physical activity and exercise training in neuromuscular diseases. Am J Phys Med Rehabil 2002; 81: S187–S219.
Raben N, Fukuda T, Gilbert AL, deJong D, et al. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from Type II muscle fibers. Mol Ther 2005; 11: 48–56.
McDonald CM . Limb contractures in progressive neuromuscular disease and the role of stretching, orthotics, and surgery. Phys Med Rehabil Clin N Am 1998; 9: 187–211.
Hyde SA, FlLytrup I, Glent S, Kroksmark AK, et al. A randomized comparative study of two methods for controlling Tendo Achilles contracture in Duchenne muscular dystrophy. Neuromuscul Disord 2000; 10: 257–263.
Ward K, Alsop C, Caulton J, Rubin C, et al. Low magnitude mechanical loading is osteogenic in children with disabling conditions. J Bone Miner Res 2004; 19: 360–369.
Tardieu C, Lespargot A, Tabary C, Bret MD . For how long must the soleus muscle be stretched each day to prevent contracture?. Dev Med Child Neurol 1988; 30: 3–10.
Jones MA, McEwen IR, Hansen L . Use of power mobility for a young child with spinal muscular atrophy. Phys Ther 2003; 83: 253–262.
Kamphoven JH, de Ruiter MM, Winkel LP, Van den Hout HM, et al. Hearing loss in infantile Pompe’s disease and determination of underlying pathology in the knockout mouse. Neurobiol Dis 2004; 16: 14–20.
Hall JW III . Handbook of Auditory Evoked Responses. Needham Heights (MA): Allyn and Bacon, 1992.
Makos MM, McComb RD, Hart MN, Bennett DR . Alpha-glucosidase deficiency and basilar artery aneurysm: report of a sibship. Ann Neurol 1987; 22: 629–633.
Matsuoka Y, Senda Y, Hirayama M, Matsui T, et al. Late-onset acid maltase deficiency associated with intracranial aneurysm. J Neurol 1988; 235: 371–373.
Anneser JM, Pongratz DE, Podskarbi T, Shin YS, et al. Mutations in the acid alpha-glucosidase gene (M. Pompe) in a patient with an unusual phenotype. Neurology 2005; 64: 368–370.
Braunsdorf WE . Fusiform aneurysm of basilar artery and ecstatic internal carotid arteries associated with glycogenosis type 2 (Pompe’s disease). Neurosurgery 1987; 21: 748–749.
Ellis FR . Inherited muscle disease. Br J Anaesth 1980; 52: 153–164.
McFarlane HJ, Soni N . Pompe’s disease and anaesthesia. Anaesthesia 1986; 41: 1219–1224.
Riou B, Lecarpentier Y, Viars P . Effects of etomidate on the cardiac papillary muscle of normal hamsters and those with cardiomyopathy. Anesthesiology 1993; 78: 83–90.
Lindeke LL, Leonard BJ, Presler B, Garwick A . Family-centered care coordination for children with special needs across multiple settings. J Pediatr Health Care 2002; 16: 290–297.
“Family Centered Care” a tutorial, 2005. Available at: http://www.communitygateway.org/faq/fcc.htm Accessed February 28, 2006.
Learning to Live with Neuromuscular Disease. A message for parents. Available at: www.mdausa.org/publications/learning/. Accessed February 28, 2006.
Hermans MM, van Leenen D, Kroos MA, Beesley CE, et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat 2004; 23: 47–56.
Besancon AM, Castelnau L, Nicolesco H, Dumez Y, et al. Prenatal diagnosis of glycogenosis type II (Pompe’s disease) using chorionic villi biopsy. Clin Genet 1985; 27: 479–482.
Park HK, Kay HH, McConkie-Rosell A, Lanman J, et al. Prenatal diagnosis of Pompe’s disease (type II glycogenosis) in chorionic villus biopsy using maltose as a substrate. Prenat Diagn 1992; 12: 169–173.
Kleijer WJ, van der Kraan M, Kroos MA, Groener JE, et al. Prenatal diagnosis of glycogen storage disease type II: enzyme assay or mutation analysis?. Pediatr Res 1995; 38: 103–106.
Van Hove JL, Yang HW, Wu JY, Brady RO, et al. High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci U S A 1996; 93: 65–70.
Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, et al. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 2000; 356: 397–398.
Van den Hout JM, Reuser AJ, de Klerk JB, Arts WF, et al. Enzyme therapy for Pompe disease with recombinant human alpha-glucosidase from rabbit milk. J Inherit Metab Dis 2001; 24: 266–274.
Amalfitano A, Bengur AR, Morse RP, Majure JM, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001; 3: 132–138.
Klinge L, Straub V, Neudorf U, Schaper J, et al. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord 2005; 15: 24–31.
Kishnani P, Spencer C, Byrne B, Nicolino M, et al. Long term efficacy of enzyme replacement therapy (ERT) in children with Pompe disease. Genet Med. In press.
Meikle P, Ranieri E, Simonsen H, Rozaklis T, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tiered strategy. Pediatrics 2004; 114: 909–916.
Van den Hout JM, Kamphoven JH, Winkel LP, Arts WE, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004; 113: e448–e457.
Winkel LP, Kamphoven JH, Van den Hout HJ, Severijnen LA, et al. Morphological changes in muscle tissue of patients with infantile Pompe’s disease receiving enzyme replacement therapy. Muscle Nerve 2003; 27: 743–751.
Kishnani PS, Nicolino M, Voit T, Rogers RC, et al. Results from a phase II trial of Chinesse hamster ovary cell–derived recombinant human acid alpha—glucosidase in Infantile—onset Pompe disease. J Pediatr 2006; in press.
Zaretsky JZ, Candotti F, Boerkoel C, Adams EM, et al. Retroviral transfer of acid alpha-glucosidase cDNA to enzyme-deficient myoblasts results in phenotypic spread of the genotypic correction by both secretion and fusion. Hum Gene Ther 1997; 8: 1555–1563.
Pauly DF, Johns DC, Matelis LA, Lawrence JH, et al. Complete correction of acid alpha-glucosidase deficiency in Pompe disease fibroblasts in vitro, and lysosomally targeted expression in neonatal rat cardiac and skeletal muscle. Gene Ther 1998; 5: 473–480.
Amalfitano A, McVie-Wylie AJ, Hu H, Dawson TL . Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci U S A 1999; 96: 8861–8866.
Martin-Touaux E, Puech JP, Chateau D, Emiliani C, et al. Muscle as a putative producer of acid alpha-glucosidase for glycogenosis type II gene therapy. Hum Mol Genet 2002; 11: 1637–1645.
Ding E, Hu H, Hodges BL, Migone F, et al. Efficacy of gene therapy for a prototypical lysosomal storage disease (GSD-II) is critically dependent on vector dose, transgene promoter, and the tissues targeted for vector transduction. Mol Ther 2002; 5: 436–446.
Fraites TJ Jr, Schleissing MR, Shanely RA, Walter GA, et al. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther 2002; 5: 571–578.
Sun B, Zhang H, Franco LM, Young SP, et al. Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther 2005; 11: 57–65.
Sun B, Zhang H, Franco LM, Brown T, et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther 2005; 11: 889–898.
Franco LM, Sun B, Yang X, Bird A, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther 2005; 12: 876–884.
Mah C, Cresawn KO, Fraites TJ, Pacak CA, et al. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther 2005.
Cresawn KO, Fraites TJ, Wasserfall C, Atkinson M, et al. Impact of humoral immune response on distribution and efficacy of recombinant adeno-associated virus-derived acid alpha-glucosidase in a model of glycogen storage disease type II. Hum Gene Ther 2005; 16: 68–80.
Bohannon RW . Reference values for extremity muscle strength obtained by hand-held dynamometry from adults aged 20 to 79 years. Arch Phys Med Rehabil 1997; 78( 1): 26–32.
Backman E, Odenrick P, Henriksson KG, Ledin T . Isometric muscle force and anthropometric values in normal children aged between 3.5 and 15 years. Scand J Rehabil Med 1989; 21( 2): 105–114.
Mathiowetz V, Wiemer DM, Federman SM . Grip and pinch strength: norms for 6- to 19-year-olds. Am J Occup Ther 1986; 40: 705–711.
Mathiowetz V, Kashman N, Volland G, Weber K, et al. Grip and pinch strength: normative data for adults. Arch Phys Med Rehabil 1985; 66: 69–74.
Piper MCaD . J Motor Assessment of the Developing Infant. Philadelphia: W B Saunders Company, 1994.
Russell D, Rosenbaum PL, Avery LM, Lane M . Gross Motor Function Measure (GMFM-66 & GMFM-88) User’s Manual. London: Mac Keith Press, 2002.
Vignos PJ Jr, Spencer GE Jr, Archibald KC . Management of progressive muscular dystrophy in childhood. Jama 1963; 184: 89–96.
Guyatt GH, Sullivan MJ, Thompson PJ, Fallon EL, et al. The 6-minute walk: a new measure of exercise capacity in patients with chronic heart failure. Can Med Assoc J 1985; 132: 919–923.
Personius KE, Pandya S, King WM, Tawil R, et al. Facioscapulohumeral dystrophy natural history study: standardization of testing procedures and reliability of measurements. The FSH DY Group. Phys Ther Mar 1994; 74: 253–263
Folio MR, Fewell RR . Peabody Developmental Motor Scales – 2, 2nd ed. San Antonio: Therapy Skill Builders, 2000.
Haley SM, Coster WJ, Ludlow LH, Haltiwanger JT, et al. The Pediatric Evaluation of Disability Inventory. Boston: Center for Rehabilitation Effectiveness, Boston University, 1992.
Haley SM, Fragala MA, Aseltine R, Ni P, Skrinar AM . Development of a disease-specific disability instrument for Pompe disease. Pediatr Rehabil 2003; 6: 77–84.
Keith RA, Granger CV, Hamilton BB, Sherwin FS . The functional independence measure: a new tool for rehabilitation. Adv Clin Rehabil 1987; 1: 6–18.
Ottenbacher KJ, Msall ME, Lyon NR, Duffy LC, et al. Interrater agreement and stability of the Functional Independence Measure for Children (WeeFIM): use in children with developmental disabilities. Arch Phys Med Rehabil 1997; 78: 1309–1315.
Krechel SW, Bildner J . CRIES: a new neonatal postoperative pain measurement score. Initial testing of validity and reliability. Paediatr Anaesth 1995; 5: 53–61.
Bieri D, Reeve RA, Champion GD, Addicoat L, et al. The Faces Pain Scale for the self-assessment of the severity of pain experienced by children: development, initial validation, and preliminary investigation for ratio scale properties. Pain 1990; 41: 139–150.
Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, et al. The Faces Pain Scale-Revised: toward a common metric in pediatric pain measurement. Pain 2001; 93: 173–183.
McCaffery M . Choosing a faces pain scale. Nursing 2002; 32: 68.
Ware JE Jr, Gandek B . Overview of the SF-36 Health Survey and the International Quality of Life Assessment (IQOLA) Project. J Clin Epidemiol 1998; 51: 903–912.
Bergner M, Bobbitt RA, Carter WB, Gilson BS . The Sickness Impact Profile: development and final revision of a health status measure. Med Care 1981; 19: 787–805.
Varni JW, Seid M, Rode CA . The PedsQL: measurement model for the pediatric quality of life inventory. Med Care 1999; 37: 126–139.
Acknowledgements
We are grateful to Rochelle Hirschhorn, MD, Gregory Carter, MD, Johan van Hove, MD, PhD, James Leonard, MD, Jennifer Li, MD, and Yuan-Tsong Chen, MD, PhD for their prepublication reviews of this guideline. This project was supported by an unrestricted educational grant from the American College of Medical Genetics Foundation and Genzyme Corporation. Neither played any direct role in the committee’s deliberations or in the development and writing of this guideline. The data on clinical trials with ERT was supported by grants from Genzyme Corp. PSK, CJT, BJB, and RDS have received research/grant support and honoraria from Genzyme Corp.; PSK, CJT, and RDS are members of the Pompe Disease Advisory Board for Genzyme Corporation. RHH is a consultant to Genzyme Corp. PSK and BJB are members of the International Pompe Disease Registry Board. JFC is CEO of Amicus Therapeutics that is developing treatments for lysosomal diseases. MSW raises funds from industry for the American College of Medical Genetics Foundation. LEC has received honoraria from Genzyme Corporation and has received research support from the Leal Foundation. If therapy for Pompe disease proves successful commercially, Duke University and inventors for the method of treatment and for some of the cell lines used to generate the enzyme (rhGAA) used in clinical trials may benefit financially pursuant to the University’s Policy on Inventions, Patents and Technology Transfer, even if those cell lines are not used in the commercialized therapy.
Author information
Authors and Affiliations
Consortia
Additional information
ACMG Practice Guideline
APPENDIX A
APPENDIX A
Motor and Functional Assessments Motor and functional assessments are recommended to establish a baseline with repeat testing at 3 – 6 month intervals for children under age five years, and annually in older children and adults, except where additional testing is clinically indicated by change in function or failure to make expected progress. The following list includes most of the major assessments but is not intended to be an exhaustive listing of all possible assessments or tools used in assessments.
Musculoskeletal
-
passive ranges of motion and measures of muscle extensibility (goniometry)
-
posture and alignment in all positions
-
spine:
-
○ flexibility, stability, posture and alignment
-
○ monitoring for kyphosis, scoliosis, lordosis
-
-
hip joint stability/integrity
-
DEXA
-
radiographs as needed for monitoring hip and spine status
Qualitative/Kinematic Assessment of compensatory movement/strategies for movement Strength - “pure” measures of muscle strength (specific tests chosen based on age and level of strength)
-
manual muscle testing (MMT)
-
quantitative muscle testing
-
○ dynamometry – different types and brands
-
○ other quantitative measures (isokinetic, isotonic, isometric)
-
Functional Measures (including functional measures of strength): (specific tests chosen based on age and purpose of functional exam)
-
Alberta Infant Motor Scales126
-
Gross Motor Function Measure (GMFM)127
-
Timed Functional Tests128
-
Six Minute Walk129
-
Functional Grades130
-
Peabody Developmental Motor Scales 2131
Measures of Disability: (measures chosen may depend on age, setting, context)
-
Pediatric Evaluation of Disability Index (PEDI)132
-
Pompe PEDI133
-
Functional Independence Measure (FIM)134
-
WeeFIM135
Pain Scales: (scales used depend on age, level of function)
-
CRIES Pain Scale136
-
Pediatric Objective Pain Scale
-
Visual Analogue Scales (VAS)
Health Related Quality of Life (HRQOL) Measures
-
Short Form 36 (SF36) Health Survey140
-
Sickness Impact Profile141
-
Center for Disease Control HRQOL (www.cdc.gov/hrqol/hrqol14_measure.htm)
-
PedsQL142
Rights and permissions
About this article
Cite this article
Kishnani, P., Steiner, R., Bali, D. et al. Pompe disease diagnosis and management guideline. Genet Med 8, 267–288 (2006). https://doi.org/10.1097/01.gim.0000218152.87434.f3
Issue Date:
DOI: https://doi.org/10.1097/01.gim.0000218152.87434.f3
Keywords
This article is cited by
-
Carrier screening for present disease prevalence and recessive genetic disorder in Taiwanese population
Journal of Human Genetics (2024)
-
Long-term safety and efficacy of cipaglucosidase alfa plus miglustat in individuals living with Pompe disease: an open-label phase I/II study (ATB200-02)
Journal of Neurology (2024)
-
Population pharmacokinetic modeling and dosing simulation of avalglucosidase alfa for selecting alternative dosing regimen in pediatric patients with late-onset pompe disease
Journal of Pharmacokinetics and Pharmacodynamics (2023)
-
Aggressive immunotherapy combined with bortezomib and rituximab for membranous nephropathy associated with enzyme replacement therapy in Pompe disease
Pediatric Nephrology (2023)
-
Development of a rapid simultaneous assay of two urinary tetrasaccharide metabolites using differential ion mobility and tandem mass spectrometry and its application to patients with glycogen storage disease (type Ib and II)
Analytical and Bioanalytical Chemistry (2023)
