
Abstract
Purpose: Sotos syndrome is a genetic disorder characterized primarily by overgrowth, developmental delay, and a characteristic facial gestalt. Defects in the NSD1 gene are present in approximately 80% of patients with Sotos syndrome. The goal of this study was to determine the incidence of NSD1 abnormalities in patients referred to a clinical laboratory for testing and to identify clinical criteria that distinguish between patients with and without NSD1 abnormalities.
Methods: Deletion or mutation analysis of the NSD1 gene was performed on 435 patients referred to our clinical genetics laboratory. Detailed clinical information was obtained on 86 patients with and without NSD1 abnormalities, and a clinical checklist was developed to help distinguish between these two groups of patients.
Results: Abnormalities of the NSD1 gene were identified in 55 patients, including 9 deletions and 46 mutations. Thus, in the clinical laboratory setting, deletions were found in 2% and mutations in 21% of samples analyzed, because not all patients had both tests. Thirty-three previously unreported mutations in the NSD1 gene were identified. Clinical features typically associated with Sotos syndrome were not found to be significantly different between individuals with and without NSD1 abnormalities. The clinical checklist developed included poor feeding, increased body mass index, and enlarged cerebral ventricles, in addition to the typical clinical features of Sotos syndrome, and was able to distinguish between the two groups with 80% sensitivity and 70% specificity.
Conclusions: The dramatic decrease in the frequency of finding NSD1 abnormalities in the clinical laboratory is likely because of the heterogeneity of the patient population. Our experience from a diagnostic laboratory can help guide clinicians in deciding for whom NSD1 genetic analysis is indicated.
Similar content being viewed by others


Main
Sotos syndrome is an overgrowth condition first described in 1964,1 and the clinical features of this syndrome have been thoroughly reviewed.2 Sotos syndrome is characterized by macrocephaly, overgrowth (with height usually significantly above the 97th percentile and typically more increased than weight), advanced bone age, and characteristic dysmorphic features including long, thin facies, a broad forehead, sparse frontoparietal hair, and down-slanted palpebral fissures. Additional medical problems may include hypotonia, feeding problems, neonatal jaundice, upper respiratory tract infections, urinary tract infections, otitis media, and conductive hearing loss. Less common features of this condition include seizures (often febrile), delayed puberty in boys, and congenital heart defects.2–8 Characteristic magnetic resonance imaging changes include dilated posterior aspects of the lateral ventricles, thinning of the corpus callosum (especially the posterior third), increased size of extracerebral fluid spaces, and midline brain anomalies.9 Speech and gross motor delays are common but may not be accurate predictors of future intellectual performance. IQs range from above average to severe mental retardation with borderline intelligence being common. Individuals with Sotos syndrome may have poor social adjustment, high levels of anxiety, and an increased incidence of hyperactivity and tantrums.10 Most cases of Sotos syndrome are sporadic, but several familial cases have been reported, indicating dominant inheritance of this condition.11–14
Mutations in the NSD1 (nuclear receptor SET domain-containing protein) gene were first identified in Japanese individuals with Sotos syndrome.15,16 These initial studies reported a 2.2 Mb deletion that included NSD1 in 57% of patients and mutations within the gene in 10%. Further studies in non-Japanese patients with Sotos syndrome have shown mutations to be more predominant, occurring in 48% to 90% of patients, and the common deletion occurring in 5% to 18% of patients.14,17,18 These studies also demonstrated the lack of NSD1 mutations or deletions in individuals with other overgrowth phenotypes.14,17,18 NSD1 mutations were described in two patients with Beckwith-Wiedemann syndrome and in several patients with Weaver syndrome.17–19 However, the features in these patients overlap extensively with Sotos syndrome. The patients with reported Beckwith-Wiedemann syndrome and NSD1 mutations most likely have Sotos syndrome,20 and it still remains unclear whether Weaver syndrome is allelic to Sotos syndrome or a distinct syndrome. Thus, NSD1 abnormalities appear to be specific for Sotos syndrome.
Several groups have attempted to elucidate genotype–phenotype correlations in Sotos syndrome in an effort to define the clinical characteristics most predictive of patients confirmed to have Sotos syndrome by NSD1 mutation. Macrocephaly, developmental delay, and the facial gestalt (frontal bossing, down-slanted palpebral fissures, and pointed chin) were reported as consistent features in all children with NSD1 mutations or deletions, but overgrowth and advanced bone age were less specific.14,18,20,21 The phenotypic differences identified in deletion-positive individuals compared with mutation positive included more severe mental retardation and less prominent overgrowth in the deletion-positive group.22 Initial reports of increased incidence of cardiac defects and renal anomalies in patients with deletions have not been confirmed in larger studies.23
It has been well established that more than 80% of patients with confirmed clinical features of Sotos syndrome have an NSD1 abnormality. Most clinicians do not have extensive experience with Sotos syndrome because of its relative rarity, and the subtle nature of the facial features make it harder to confirm or dismiss the diagnosis. A diagnostic genetic test is therefore clinically useful for confirmation of the clinical suspicion or diagnosis. In this context, patients with a much broader phenotypic range are referred for testing, and the mutation detection rate in a clinical laboratory setting may be expected to be lower than that reported in patients with a confirmed clinical diagnosis. We have established clinical testing for both deletions of the NSD1 gene, by fluorescent in situ hybridization (FISH) analysis, and for mutations within the NSD1 gene, by denaturing high performance liquid chromatography (DHPLC) and targeted sequencing. Here we present an unselected population of patients whose samples were sent to our laboratory for NSD1 analysis. Within this cohort, we determined the frequency and types of mutations and defined genotype–phenotype correlations to develop recommendations for clinical laboratory testing when Sotos syndrome is suspected.
MATERIALS AND METHODS
Patient samples
Patient samples received in our clinical cytogenetics and molecular genetics laboratories for NSD1 deletion and mutation analyses were included in this study after approved institutional review board protocols. Referring physicians were asked to complete a clinical questionnaire on their patients. The form included questions about pregnancy complications, growth parameters at birth, neonatal complications, developmental progress, medical complications, current growth parameters, dysmorphism (specifically with regard to Sotos facial gestalt, including frontal bossing and downslanting palpebral fissures), other physical examination findings, the results of other genetic testing, magnetic resonance imaging and computed tomography results, bone age results, family history, and ethnicity. Height/length, weight, head circumference (occipitofrontal circumference), and body mass index (BMI) were plotted on sex- and age-appropriate CDC growth charts24 to obtain percentiles.
Genotype–phenotype correlations were performed only on samples for which the clinical questionnaire was available and for which a deletion or mutation was identified or both deletion and mutation analyses were negative.
FISH analysis
PAC clone RP1-118M12, corresponding to the NSD1 gene, was used for Sotos syndrome deletion analysis. This clone was previously reported as part of the identification of haploinsufficiency of NSD1 as a cause of Sotos syndrome.15 DNA was isolated using an automated DNA isolation system (AutoGen 740, Integrated Separation Systems, Holliston, MA) and directly labeled with Spectrum Orange-dUTP (Vysis/Abbott, Inc., Downers Grove, IL), using a standard nick-translation reaction. A control probe for the 5q telomere region, RP1-240G13,25 was directly labeled with Spectrum Green-dUTP (Vysis/Abbott, Inc.) and included for dual-color FISH analysis.
Slide preparation, probe preparation, and hybridization were completed using previously described methods.26 Slides were washed in 0.4× standard saline citrate (SSC)/0.3% NP-40 for 2 minutes at 73°C, then transferred into 2× SSC/0.1% NP-40 at room temperature for 2 minutes. Slides were briefly rinsed in 4× SSC/0.1% Tween-20 at room temperature and then mounted in antifade solution (Vector, Burlingame, CA) containing DAPI.
Metaphase FISH analysis on cells from healthy control individuals was used to verify the cytogenetic position and unique localization of the NSD1 clone, and cells from an individual with a known deletion of the NSD1 gene were used for validation purposes. At least 10 cells per individual were analyzed using direct microscopic visualization and digital-imaging analysis (ViewPoint software, Vysis, Inc.).
Mutation analysis
DNA was isolated from patient peripheral blood samples using the Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN), following the manufacturer's recommendations. The coding region of the NSD1 gene (exons 2-23) and flanking intron sequences were targeted for mutation analysis. Mutation analysis was performed by a combination of DHPLC and direct sequencing methods. Exons 13, 14, and 18 were analyzed by direct sequencing because of unfavorable melting characteristics making them unsuitable for DHPLC analysis, and portions of exons 5 and 23 were sequenced because of the presence of several common polymorphisms. The remaining exons were subject to mutation analysis by DHPLC. The WAVEMAKER 4.1 software (Transgenomic Inc., Omaha, NE) was used for fragment design and selection of the optimal denaturing temperatures for DHPLC analysis. GC clamps were added to primers when necessary to obtain fragments with more uniform melting behavior. Primers were designed using the publicly available software programs, OLIGO (http://www.hitachisoft-bio.com/products/oligo/default.htm) and Primer 3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi).
The polymerase chain reaction (PCR) amplicon length ranged from 188 to 665 bp. PCR reactions for DHPLC analysis contained 2.5 mM MgCl2, 200 μM each dNTP, 10 pmol of each primer, 1 U AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA) and 100 ng of DNA in a final volume of 50 μL. PCR reactions for sequence analysis were the same except scaled down to a final volume of 25 μL. The PCR cycling conditions were as follows: 94°C for 10 minutes, followed by a touchdown cycling procedure with denaturation at 94°C for 45 seconds, annealing at 66°C for 30 seconds, decreasing by 0.5°C per cycle, and extension at 72°C for 45 seconds, for a total of 14 cycles. The amplification was continued as above for a further 25 cycles at a constant annealing temperature of 59°C followed by a final extension at 72°C for 10 minutes. The PCR products were denatured at 94°C for 5 minutes and cooled to 45°C at a rate of 0.5°C per 36 seconds for heteroduplex formation before DHPLC analysis. DHPLC analysis was performed using the WAVE 3500HT DNA fragment analysis system (Transgenomic Inc.). Each PCR product was analyzed at 50°C for sizing purposes and three predetermined optimal temperatures for denaturation and mutation detection purposes. PCR product (7 μL) was analyzed at each temperature on a DNASep HT column (Transgenomic Inc.) using the appropriate acetonitrile gradient range over 3.8 minutes. Denaturing profiles for each amplicon were always compared with a normal control sample and a sample with a known sequence change when available. Any sample with a variation in the DHPLC denaturing profile was sequenced for that particular amplicon to determine the underlying sequence change. Our DHPLC mutation assay was validated by analyzing a series of normal control samples and 26 samples with a known NSD1 sequence status, including nine blinded samples
Double-stranded DNA sequence analysis was performed using the Big Dye Terminator chemistry (Applied Biosystems), and reactions were run on the ABI 3100 Genetic Analyzer (Applied Biosystems). Sequence chromatograms were analyzed using the Sequencher software version 4.1 (GeneCodes Inc., Ann Arbor, MI), and the sequence was compared with that obtained from a normal control sample and the reference sequence for NSD1 (Genbank accession number AF395588).
Real-time quantitative polymerase chain reaction
Deletion analysis for 10 patient samples was performed by real-time quantitative PCR because only DNA was available. A portion of exon 5 of the NSD1 gene was amplified using primers 5′:-GTGACATAGAAACAGCAGTGGTGA-3′: (forward primer) and 5′:-GATGGCTTTGATGTTCCAGAG-3′: (reverse primer) and quantified by comparison with an internal control gene, albumin, amplified using primers 5′:-AATGCTGCACAGAATCCTTGGT-3′: (forward primer) and 5′:-TCATCGACTTCCAGAGCTGAAA-3′: (reverse primer). The amplifications were performed in a 50 μL reaction volume containing 25 μL SYBR Green PCR Master Mix (Applied Biosystems), 0.5 μM each primer, and 80 ng DNA. Two normal controls and two positive controls (confirmed NSD1 deletions) were included. Samples were amplified in duplicate for both NSD1 and albumin. The PCR thermal cycling conditions were as follows: 50°C for 2 minutes and 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Standard curves for both NSD1 and albumin were obtained using serial DNA dilutions of a normal control sample. Absolute quantification of target amplicons was performed by interpolation of the threshold cycle number (Ct) against the corresponding standard curve. Quantitative data were normalized against a diploid reference gene (ratio NSD1: albumin). Normalized values of 0.8 to 1.2 indicate a diploid situation; values of 0.4 to 0.6 indicate a deletion.
Statistical analysis
The two groups of patients with a genetic change (either deletion or mutation) and with no genetic change were compared through the collected variables. Most variables were binary (i.e., presence/absence indicator), and the measurements were transformed to 95% and 97% percentiles. Variables were compared between groups using the Fisher exact test for independence. Logistic regression analysis was used on each feature to develop a weighted coefficient for each variable with statistical significance and a formula to predict a probability of genetic change given the observed data. The small sample number and missing data did not allow the logistic model to fit well. The checklist was generated using the features suggested by the Fisher exact test and logistic regression analysis. An exhaustive search of all tables using these features and assigning a point value of 1 to 3 for each feature was performed. Each patient was scored as a percentage of the total score. For missing data, a patient's percentage was the point total of the total possible for the clinical information available. The Youden index was used to find the best cutoff score for each generated checklist. The Youden index is defined as the sum of specificity and sensitivity minus 1. The final checklist and cutoff were selected to maximize the Youden index over all checklists and cutoffs.
RESULTS
Initial deletion and mutation analyses were performed on 15 patients seen by one of the authors (G.B.S.) and confirmed to have a clinical diagnosis of classic Sotos syndrome. Of these clinically confirmed patients, 12 of 15 (80%) had a detectable NSD1 alteration with two deletions (13%) and 10 mutations (67%), a mutation rate consistent with previous reports.
Over the past 30 months, our laboratory has received 435 patient samples for NSD1 analysis. Of these patient samples, deletion and mutation analyses were requested on 160 (37%), deletion analysis only was requested on 218 (50%), and mutation analysis only was requested on 57 (13%). Of the 378 patients who had FISH testing for deletion analysis, 9 deletions were detected, resulting in a 2% deletion detection rate. Of the 217 patients who had mutation analysis, 46 patients had a mutation detected, and 3 patients had variants of unknown significance, with a combined mutation detection rate of 23%.
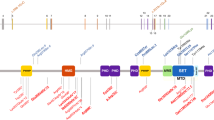
A wide range of mutations was identified, including nonsense, frameshift, splice site, and missense mutations, similar to previous reports (Table 1). Thirty-three of the mutations identified have not been previously described, including a duplication and an insertion/deletion mutation. Approximately half of the point mutations identified (nonsense and missense mutations) occurred at CpG sites. Protein truncating mutations, such as nonsense, insertion, deletion, and splice site mutations, were found to be scattered throughout the gene with the majority in exons 5, 19, 22, and 23, whereas missense mutations were found predominantly in those exons that code for known functional domains (Fig. 1).

Genomic structure of NSD1 indicating the locations of mutations identified. Known functional domains in NSD1 as previously described (shaded areas).17
Missense mutations were considered to be pathogenic if they resulted in nonconservative amino acid changes in functional domains. Many of the missense changes were in exons where other missense mutations have been described.17,18 Sequence changes were considered as variants of unknown significance when they resulted in a conservative amino acid change in a region of the gene not known to code for a functional domain and when parental samples were not available to determine whether the sequence change was de novo. In Case 47, two missense changes were identified, and it is possible that one of these changes is pathogenic. Parental samples were not available to determine whether these changes were inherited or de novo in the patient. The variants of unknown significance were checked for exon splice enhancer (ESE) motifs using the ESE finder software27 in an effort to determine whether any of these changes might affect a potential ESE that would be predicted to affect normal splicing. In patient 47, the 4363A>G sequence change appears to affect a putative ESE, and in patient 48, the single missense change 1811G>T appears to create two putative ESEs. These are merely predictions, however, and have not been confirmed by RNA studies. Two missense amino acid sequence changes were also identified in patient 53 (Table 1). Parental samples were available that demonstrated that the sequence changes were inherited from each parent. Because the parents were both reportedly healthy, we concluded that both these amino acid changes are likely to be polymorphic.
The NSD1 gene has many described polymorphisms. Several new polymorphisms were identified in this study (Table 1). These genetic changes were considered probable polymorphisms if they were found in a patient with a mutation, were found in an unaffected parent, or did not result in an amino acid change. The sequence changes that did not result in an amino acid change, that were present as the only sequence change in the patient, and in whom parental samples were not available for testing should be interpreted with caution, however, because they may potentially affect splicing if they affect ESE sequences.28
The majority of the NSD1 mutations identified were unique. Three mutations were found to occur more than once. The R1322X mutation was found in patients 5 and 6 and one previously described patient.29 The Q1989X mutation was found in patients 10 and 11, and the R1984Q mutation was found in patients 36, 37, 38, and 39, and one previously described patient.18 Parental samples were available for patients 36 and 39, and the R1984Q mutation was determined to be de novo in both cases, further supporting the pathogenic nature of this mutation.
Three of the NSD1 mutations were found to be familial. In each case, the clinicians reported a suspicion of Sotos syndrome in the affected parent before the testing. The father of patient 3, with a R1023X mutation, carried the same mutation, whereas the mother of patient 26, with a 6291delG mutation, also had the same mutation. Patient 34, with a R1663C mutation, had a father and two siblings reported to have clinical features of Sotos, and all were found to have the same mutation. An additional sibling with normal testing results did not have the mutation.
The clinical features checklist was initially developed from 60 patients. Twenty-three of the patients had an abnormality of NSD1 (7 with a deletion and 16 with a mutation), and 37 had normal deletion and mutation studies. The clinical features of these patients were compared by dividing the group into those with a discovered NSD1 abnormality and those without one. The Fisher exact test in the original group of 60 patients showed significance for poor feeding, “Sotos gestalt,” large hands and feet, height greater than the 95th percentile, and hydrocephalus/enlarged ventricles. Logistic regression analysis showed a negative association for increased weight and BMI and presence of a genetic abnormality. The small number of patients and some missing variables did not allow the logistic model to fit well. A fixed checklist was developed with an individual score defined as the percentage of the highest possible sum for all variables. The diagnostic variables for the fixed checklist included poor feeding, Sotos gestalt, length greater than or equal to the 95th percentile, large hands and feet, and hydrocephalus/enlarged ventricles according to the Fisher exact test and BMI according to the logistic regression analysis. BMI association was reversed giving a positive score if the patient's BMI was below the 95th percentile and a 0 score if equal to or above the 95th percentile. Macrocephaly, defined as occipitofrontal circumference greater than or equal to the 97th percentile, was included, because this is a common feature of Sotos syndrome. Each variable was considered with a point value of 1 to 3. An exhaustive search through all possible checklists with different point values for each variable was computed, and the final checklist and cutoff were selected to maximize the Youden index.
Figure 2 shows the final, fixed, scored checklist with performance on the first 60 patients. The total score for each patient was calculated as a percentage of the score divided by total possible points given the information available (because some data were missing). With the shown point values and a cutoff of 50%, the checklist distinguished the patients with and without NSD1 abnormality with a sensitivity of 82.6% and a specificity of 75.6%.

List of features and weighted score for the clinical checklist. Graph shows the distribution of the first 60 patients' scores between those with and without NSD1 alterations. The total score for each patient was calculated as a percentage of positive features divided by number of features for which information was available. Scores shown as percentages in graph. BMI, body mass index.
An additional 34 patients with complete clinical information and NSD1 data were used to validate the checklist. This group of patients contained 1 deletion, 12 mutations, and 21 with no mutations or deletions. Of the 13 patients with NSD1 alterations, 11 had a score greater than 50% and two had a score of less than 50%, giving a sensitivity of 85%. For the patients with normal testing, eight had a score of greater than 50% and 13 had a score of 50% or less. Overall, when the two cohorts were combined, the checklist resulted in a sensitivity of 83% and a specificity of 70%.
The clinical features of the patients with deletion, mutation, or normal NSD1 analysis are presented in Table 2. The P values from the Fisher exact test comparing the two groups (combining patients with deletions and mutations) for the entire group of patients is included.
DISCUSSION
Sotos syndrome is a well-described genetic disorder characterized clinically by overgrowth, characteristic facial features, developmental delay, and a host of medical complications. Until recently, the diagnosis was established on a clinical basis by recognition of these features. Given that Sotos syndrome is relatively rare, most clinicians have limited experience with the condition, and there is significant overlap with other overgrowth conditions. Given these complicating factors, confirmation of the clinical diagnosis can be difficult.
Identification of mutations in the NSD1 gene as causative of Sotos syndrome has allowed independent confirmation of the condition when clinically suspected. Several reports have now been published describing the frequency and type of NSD1 mutations in patients clinically diagnosed with classic Sotos, Sotos-like phenotype, Weaver syndrome, and other overgrowth conditions. Approximately 70% to 80% of patients with a clinical diagnosis of Sotos syndrome have an NSD1 abnormality.14,17,18 Most of these reports have focused on describing the specific characteristics of the patients with NSD1 mutations, further defining the specific features diagnostic of Sotos syndrome. The facial features and macrocephaly characterize the “Sotos gestalt,” which is reported to be the best indicator of finding a mutation in NSD1.14,17,18,20–22
Clinical laboratory testing is useful in confirming a suspicion of Sotos syndrome and is performed by FISH or real-time quantitative PCR for deletion analysis and DHPLC/sequencing for mutation analysis. In the clinical laboratory setting, the features indicative of Sotos syndrome in the patients referred for NSD1 analysis are not always apparent or clear. In our cohort of patients referred for NSD1 analysis, the incidence of abnormalities was 25%, which is significantly lower than the published 70% to 80% in patients clinically diagnosed with Sotos syndrome. This finding indicates that the patient cohort undergoing testing is much broader and not necessarily defined by the classic clinical characteristics of Sotos syndrome. We elected to study the clinical features of the patients referred to our laboratory for testing and compare the features of NSD1 mutation-positive and negative patients to see whether there were recognizable differences between the two groups.
The clinical checklist shown in Figure 2 was developed to maximize the sensitivity based on the clinical features shown in Table 2. Given that NSD1 mutations are expected in 70% to 80% of patients with Sotos syndrome, no specific clinical feature or checklist will be perfect, because 20% of patients with positive clinical findings will not have an NSD1 mutation. With a cutoff of greater than 50% for the total possible score, our checklist was able to predict those patients with an NSD1 mutation with approximately 80% sensitivity and 70% specificity. The main distinguishing features in our checklist were poor feeding in the newborn period, dilated ventricles or hydrocephalus, and BMI less than the 95th percentile. A higher incidence of feeding problems was noted in another study,13 which may be due in part to the high incidence of hypotonia in the neonatal period. Hydrocephalus is not a significantly common feature of Sotos syndrome. However, the combination of macrocranium without megalencephaly9 produces enlarged ventricles and increased size of the extra-axial spaces, a combination oftentimes mistaken for “communicating hydrocephalus.” In the clinical survey the feature “enlarged ventricles/hydrocephalus” was reported in the patients with NSD1 mutations more frequently than that of the NSD1-negative group. Many of the NSD1-negative patients had an increased BMI consistent with significant obesity. Obesity is not a common feature of Sotos syndrome but was reported in more than half the patients who were negative for NSD1 mutations.
The checklist also included macrocephaly, facial gestalt, and height greater than the 95th percentile, because these features had been suggested by other studies to be characteristic for patients with NSD1 mutations.7–11 These features were also seen commonly in the group of patients without an identifiable NSD1 mutation. The NSD1-negative group had macrocephaly and tall stature in 86% and 78%, respectively, compared with the NSD1-positive group in whom these features were present in 83% and 94%.
The characteristic dysmorphic features defined in our questionnaire as “Sotos facial gestalt (e.g., frontal bossing and downslanting palpebral fissures)” were reported to be present in 97% of the mutation-positive group, as would be expected. Surprisingly, this feature was also reported as present in 80% of the mutation-negative group. The only other published, similar scored checklist is based on the dysmorphic features of downslanted palpebral fissures, pointed chin, frontal bossing, and large growth.21 The dysmorphic features are subtle, and recognition of the overall gestalt is most likely aided by the experience of examining many affected patients. The authors of previous articles have the advantage of reviewing or personally examining many patients with Sotos syndrome, making it easier to recognize the dysmorphic features and distinguish the patients who are affected. The clinicians referring patient samples to our laboratory most likely will not have seen a large cohort of patients with Sotos syndrome. This lack of experience, as well as variability in dysmorphology training, may well explain the lack of specificity of the Sotos facial gestalt.
Individuals with the clinical manifestations of Sotos syndrome are brought to the attention of neurologists, developmental pediatricians, cardiologists, and medical geneticists. In our set of patients, the majority of cases were referred by geneticists (87%), and a minority from neurologists (9%) and other specialists including developmental pediatricians (4%). The high percentage of NSD1-negative patients reported with the facial features of Sotos syndrome cannot be completely explained by referrals from clinicians with a lack of formal training in dysmorphology.
There were differences between the groups in rate of diagnosis and pattern of tests requested. The geneticists had a positive mutation detection rate of 14% compared with 3% and 6% for neurologists and other physician/specialist groups. NSD1 genetic testing includes deletion and mutation analyses, which should be positive in 10% to 15% and 60% to 70% of cases, respectively. Geneticists ordered deletion analysis alone in 44% of the patients, mutation analysis alone in 16%, and both tests in 40% of patients. In contrast, the neurologists ordered deletion analysis alone in 89%, mutation analysis only in 2%, and both in 9% of patients, whereas the remaining physicians ordered deletion analysis alone in 69%, mutational analysis alone in 18%, and both in 13% of patients. The reasons for these testing patterns are not clear. Geneticists may be more aware of the mutational spectrum, because most of the publications regarding NSD1 and Sotos syndrome to date have been in genetics journals. The additional cost of DHPLC/sequencing analysis versus a single FISH assay may also come into consideration. The testing pattern cannot explain the overall low mutational detection rate in these patients, however, further implicating the ability to recognize the clinical phenotype.
There were a total of six patients (two deletions and four mutations) with an identified NSD1 alteration who were at or below the 50% score cutoff on the checklist. Of these, one patient with mutation had a score of 6 (50%), with data missing on two of the variables. One patient with deletion with a score of 4 (33%) had a head circumference at the 85th percentile and height at the 50th percentile and was also missing two pieces of data. Patients with deletions have been reported to have less overgrowth compared with patients with mutations,22 which will complicate the recognition of these patients. The missing data may explain why these two patients scored below the cutoff on the checklist, and both patients may have an increased score if all data were available. The second patient with deletion had a score of 6 (50%), which included poor feeding, Sotos gestalt, macrocephaly, and advanced height, but BMI was slightly higher than the 95th percentile. Despite having overgrowth, the patient's hands and feet were not reported as large, although exact measurements were not available. Two patients with mutations had a score of four (large hands/feet, Sotos gestalt, macrocephaly, advanced height) but did not have any of the other features. These two patients indicate one of the drawbacks of the checklist. The heavily weighted features are not the typical ones given in the previous clinical descriptions of Sotos syndrome. These features are weighted on the basis of the comparison between the groups in this patient cohort. These two patients with the apparent clinical diagnosis do not score above 50% because of their lack of the heavily weighted less-than-common features.
Finally, one of the patients with a score of 3 (25%) had an interesting, unique familial mutation R1663C (Table 1). The patient, his father, and two of his male siblings were also found to have the mutation, but the mother and one male sibling did not carry the sequence change. Clinical information was available only on two of the affected siblings and one healthy sibling. The affected siblings had macrocephaly but were at the 75th to 80th percentile for height. One had large hands and feet, and the other did not. Neither had increased BMI, poor feeding, or large ventricles. Both affected brothers and the nonaffected brother, as well as the father, were reported to have the facial gestalt, highlighting the difficulty of recognizing the true Sotos syndrome facial features. The unaffected sibling's height was at the 25th percentile. This family's mutation has not been reported, and the main features suggestive of Sotos syndrome were macrocephaly and the dysmorphic features. Overgrowth was not a significant feature in this family. This unique mutation may lead to a slightly different phenotype, more reminiscent of patients with deletions who have less prominent overgrowth than patients with mutations.22
The mutational spectrum reported in these patients is similar to the pattern of mutations previously described and includes 33 novel mutations. One mutation, R1984Q, was identified in four unrelated patients, accounting for 9% of the mutations identified in our patient population. The de novo nature of the R1984Q mutation in two of our patients, as well as in a previously described patient,18 demonstrates that this mutation has arisen separately and may represent a mutation hot spot. The R1984Q mutation does occur at a hypermutable CpG site that may be the basis of its recurrence. The ethnicity of three of the four patients was white in one patient and mixed Cherokee Indian in two patients. The R1984Q mutation represents the most common mutation identified in the NSD1 gene to date.
Three familial cases were found, indicating that recurrence in families may be more common than previously thought, although the overall incidence remains low. Two of the three mutations were protein truncating, whereas one was a missense mutation in a known functional domain. The familial mutations identified, therefore, do not appear to be less severe compared with mutations in the sporadic cases.
The diagnosis of Sotos syndrome can now be confirmed by genetic testing of NSD1 but remains challenging from a clinical basis. Several studies have now been reported that further clarify and specify the clinical features diagnostic of Sotos syndrome. These reports are detailed descriptions of the features of patients with known NSD1 mutations. Many of the features, however, are not specific for Sotos syndrome and can be seen in a large number of patients with other conditions. The only feature that seems to be specific for Sotos syndrome is the dysmorphic facies, but as our study suggests, this gestalt is difficult to recognize by clinicians, including medical geneticists. In this study, feeding difficulties were recognized to be more common in Sotos syndrome, which is in agreement with other reports.21 In addition, obesity was a negative predictor for finding an NSD1 mutation.
CONCLUSIONS
To date, NSD1 abnormalities have been found only in patients with recognized Sotos syndrome and have not been implicated in patients with other overgrowth conditions or in patients with only overgrowth or macrocephaly. Therefore, NSD1 analysis is less useful in these latter groups of patients. No set of criteria or checklists have been 100% accurate in predicting which patients will have an NSD1 abnormality, so careful attention to all of the reported features is important. The checklist we have developed should provide a useful guide when considering NSD1 analysis. Finally, it is important to keep in mind that 20% of patients with unambiguous Sotos syndrome will not have a detectable abnormality, and patients with deletions may not have overgrowth as a prominent feature. Familial mutations are not as rare as previously thought. This will affect recurrence risk counseling and demands close examination of the parents. As with other rare conditions, access to pictures of many affected children will help the clinician to obtain more familiarity with the condition and aid in the diagnosis and appropriate use of genetic testing.
References
Sotos JF, Dodge P, Muirhead D, Crawford J, Talbot N . Cerebral gigantism in childhood. A syndrome of excessively rapid growth with acromegalic features and a nonprogressive neurologic disorder. N Engl J Med 1964; 271: 109–116.
Cole TRP, Hughes HE . Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31: 20–32.
Evans PR . Sotos' syndrome (cerebral gigantism) with peripheral dysostosis. Arch Dis Child 1971; 46: 199–202.
Bale AE, Drum MA, Parry DM, Mulvihill JJ . Familial Sotos syndrome (cerebral gigantism): craniofacial and psychological characteristics. Am J Med Genet 1985; 20: 613–624.
Wit JM, Beemer FA, Barth PG, Oorthuys JW et al. Cerebral gigantism (Sotos syndrome). Compiled data of 22 cases. Analysis of clinical features, growth and plasma somatomedin. Eur J Pediatr 1985; 144: 131–140.
Kaneko H, Tsukahara M, Tachibana H, Kurashige H, Kuwano A, Kajii T . Congenital heart defects in Sotos sequence. Am J Med Genet 1987; 26: 569–576.
Goldstein DJ, Ward RE, Moore E, Fremion AS, Wappner RS . Overgrowth, congenital hypotonia, nystagmus, strabismus, and mental retardation: variant of dominantly inherited Sotos sequence?. Am J Med Genet 1988; 29: 783–792.
Buyukgebiz A, Kinik E . Sotos syndrome presenting with epilepsy. Turk J Pediatr 1990; 32: 59–63.
Schaefer GB, Bodensteiner JB, Buehler BA, Lin A, Cole TRP . The neuroimaging findings in Sotos syndrome. Am J Med Genet 1997; 68: 462–465.
Sarimski K . Behavioral and emotional characteristics in children with Sotos syndrome and learning disabilities. Dev Med Child Neurol 2003; 45: 172–178.
Cole TR, Hughes HE . Sotos syndrome. J Med Genet 1990; 27: 571–576.
Opitz JM, Weaver DW, Reynolds JF Jr . The syndromes of Sotos and Weaver: reports and review. Am J Med Genet 1998; 79: 294–304.
Hoglund P, Kurotaki N, Kytola S, Miyake N, Somer M, Matsumoto N . Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet 2003; 40: 51–54.
Turkman S, Gillessen-Kaesbach G, Meinecke P, Albrecht B et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 2003; 11: 858–865.
Kurotaki N, Imaizumi K, Harada N, Masuno M et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30: 365–366.
Kurotaki N, Harada N, Shimokawa O, Miyake N et al. Fifty microdeletions among 112 cases of Sotos syndrome: low copy repeats possibly mediate the common deletion. Hum Mutat 2003; 22: 378–387.
Douglas J, Hanks S, Temple IK, Davies S et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Rio M, Clech L, Amiel J, Faivre L et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet 2003; 40: 436–440.
Baujat G, Rio M, Rossignol S, Sanlaville D et al. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet 2004; 74: 715–720.
Tatton-Brown K, Rahman N . Clinical features of NSD1 positive Sotos syndrome. Clin Dysmorphol 2004; 13: 199–204.
de Boer L, Kant SG, Karperien M, van Beers L et al. Genotype-phenotype correlation in patients suspected of having Sotos syndrome. Horm Res 2004; 62: 197–207.
Nagai T, Matsumoto N, Kurotake N, Harada N et al. Sotos syndrome and haploinsufficiency of NSD1: clinical features of intragenic mutations and submicroscopic deletions. J Med Genet 2003; 40: 285–289.
Tatton-Brown K, Douglas J, Coleman K, Baujat G et al. Multiple mechanisms are implicated in the generation of 5q35 microdeletions in Sotos syndrome. J Med Genet 2005; 42: 307–313.
2000 Centers for Disease Control Growth Charts: United States. Available at: http://www.cdc.gov/growthcharts/ Accessed January 30, 2005.
Knight SJL, Lese CM, Precht K, Kuc J et al. An optimized set of human telomere clones for studying telomere integrity and architecture. Am J Hum Genet 2000; 67: 320–32.
Chong SS, Pack SD, Roschke AV, Tanigami A et al. A revision of the lissencephaly and Miller-Dieker syndrome critical regions in chromosome 17p13.3. Hum Mol Genet 1997; 6: 147–155.
ESE finder. Available at: http://rulai.cshl.edu/tools/ESE/. Accessed February 9, 2005.
Cartegni L, Chew SL, Krainer AR . Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev 2002; 3: 285–298.
Kamimura J, Endo Y, Kurotaki N, Kinoshita A et al. Identification of eight novel NSD1 mutations in Sotos syndrome. J Med Genet 2003; 40: e126.
Acknowledgements
We thank Dr. Nazneen Rahman at the Institute of Cancer Research, Surrey, United Kingdom, who provided the blinded samples, and all the clinicians who provided the clinical questionnaire.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Waggoner, D., Raca, G., Welch, K. et al. NSD1 analysis for Sotos syndrome: Insights and perspectives from the clinical laboratory. Genet Med 7, 524–533 (2005). https://doi.org/10.1097/01.GIM.0000178503.15559.d3
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000178503.15559.d3
Keywords
This article is cited by
-
Drosophila NSD deletion induces developmental anomalies similar to those seen in Sotos syndrome 1 patients
Genes & Genomics (2021)
-
A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection
Human Genome Variation (2017)
-
Cooperative genetic defects in TLX3 rearranged pediatric T-ALL
Leukemia (2008)
-
Mutation analysis of the NSD1 gene in patients with autism spectrum disorders and macrocephaly
BMC Medical Genetics (2007)
