
Abstract
Purpose Generalized atrophic benign epidermolysis bullosa is a nonlethal form of junctional EB with an autosomal recessive inheritance. There is generalized cutaneous blister formation at sites of trauma, atrophic alopecia affecting scalp, eyelash and eyebrow, dystrophic nail changes, and tooth abnormalities. In this study, we have studied a five-generation Palestinian family affected with generalized atrophic benign epidermolysis bullosa.
Methods We have performed linkage analysis to genes that are mutated in generalized atrophic benign epidermolysis bullosa, followed by direct sequencing of patient genomic DNA.
Results We have shown that the disease is caused by a newly detected homozygous donor splice site mutation, IVS51+1G>A, in the type XVII collagen gene, COL17A1.
Conclusion The effect of a founder mutation introduced 3 to 4 generations before a disease appearance is demonstrated in this inbred family.
Similar content being viewed by others


Main
Epidermolysis bullosa (EB) is a term given to a group of mechanobullous disorders characterized by fragility of the skin and mucous membranes after mechanical trauma. There are over twenty types of EB but they can be grouped into three main subtypes based on the level of cleavage within the skin. 1 Generalized atrophic benign EB (GABEB)(MIM 226650), also known as non-Herlitz junctional EB, is a late onset autosomal recessive form of junctional EB. Junctional types of EB (JEB) are characterized by dermal-epidermal separation through the lamina lucida of the basement membrane. 2 Although GABEB generally has a good prognosis, 3 some cases have been shown to be associated with chronic renal failure. 4 Clinically there is generalized cutaneous blister formation at sites of trauma, atrophic alopecia affecting scalp, eyelash, and eyebrow, dystrophic nail changes, tooth abnormalities, and sometimes oral mucosal lesions. 5 The blisters on the skin and mucous membranes heal without scarring or dystrophy but often result in notable atrophy 3,6,7 and some patients develop squamous cell carcinomas at sites of chronic wound healing. 8
Immunohistochemical analysis of lesional skin reveals deficient expression of either type XVII collagen or laminin-5, 9–11 thus demonstrating the genetic heterogeneity of GABEB. This genetic heterogeneity was confirmed by the discovery of mutations in the genes encoding either type XVII collagen, COL17A112 or the laminin 5 peptides, LAMB3, 11,13LAMA3, 14 and LAMC2. 14
The type XVII collagen gene (COL17A1) previously known as 180-kD bullous pemphigoid antigen (BPAG2) comprises 56 distinct exons, which span approximately 52 kb of chromosome 10q24.3. 15,16 The gene encodes an α1(XVII) chain that consists of an intracellular globular domain, a transmembrane segment, and a collagenous extracellular domain. The extracellular domain contains 15 subdomains 15 to 242 amino acids in length composed of Gly-X-Y tandem repeats that are predicted to form collagen-like triple helices. 15–19 The carboxyl terminus of type XVII collagen is localized within the lamina densa, extending from the intracellular hemidesmosomal plaque of basal keratinocytes to the lamina densa of the epidermal basement membrane. 20
In this study, we have studied a large family of Palestinian Arab origin with GABEB and have demonstrated linkage of the disease to the type XVII collagen gene, COL17A1. We have shown that the disease is caused by a donor splice site mutation in intron 51 introduced by a founder individual 3 generations before disease appearance. A high rate of consanguinity has led to the birth of six affected patients in the fifth generation.
METHODS
Clinical
The family was seen by a dermatologist (I.G.) and geneticists (O.R. and C.S.) at Assaf Harofeh Medical center, Israel. Informed consent was obtained from the parents and the guardians, and genomic DNA was extracted from blood samples using standard methods.
Linkage analysis
Due to the genetic heterogeneity of GABEB, microsatellite markers proximal and distal to the LAMA3, LAMB3, LAMC2, and COL17A1 genes were amplified in a 7.5-μL mix containing 40 ng genomic DNA, 5 pmol of forward and reverse primers, 2.5 nmol each dNTP, 10 mmol/L Tris-HCl, 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.25 U Amplitaq Gold Taq polymerase (Applied Biosystems). See Table 1 for the markers used. The primer sequences are available from the Genome Database (http://www.gdb.org/). Forward or reverse primers were labeled with 6-FAM, or HEX fluorescent dyes. Reaction conditions were initial denaturation of 95°C for 12 minutes, followed by 10 cycles of 95°C for 30 seconds, 52°C for 30 seconds, 72°C for 1 minute, and then 30 cycles of 89°C for 30 seconds, 52°C for 30 seconds, and 72°C for 1 minute, with a final step of 72°C for 10 minutes. PCR products were separated by polyacryamide gel electrophoresis on an ABI 377 DNA sequencer and analyzed using Genescan 3.0 and Genotyper 2.5 software (Applied Biosystems, Warrington, UK).
Mutation detection in the COL17A1 gene
Individual exons and splice sites of the COL17A1 gene were amplified using primers based on the genomic sequence (http://genome.ucsc.edu/). Specifically, exon 51 and its splice sites were amplified using primers COL17A1 51F (forward) 5′-GATGATGTCAGGAAACAGCAG-3′, and COL17A1 51R (reverse) 5′-TCGTGTGGCCTTCCTGTCC-3′. Briefly, PCR products were amplified using 50 ng genomic DNA in a 50 μL mix containing 20 pmol forward and reverse primers, 10 nmol each of dTTP, dCTP, and dATP, 7.6 nmol dGTP, 2.5 nmol deaza GTP, 2.5 μL DMSO, 50 μmol betaine, 10 mmol/L Tris-HCl, 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 1.0 U Amplitaq Gold Taq polymerase (Applied Biosystems, Warrington, UK). After an initial denaturation of 95°C for 12 minutes, 40 cycles were performed of 95°C for 10 seconds, 55°C for 30 seconds, and 72°C for 1 minutes, with a final extension step of 72°C for 10 minutes. The PCR products were examined by 2% agarose gel electrophoresis, purified using spin columns (Qiagen, Crawley, UK) and sequenced using a Big Dye terminator kit on an ABI 377 genetic analyzer (ABI, Foster City, California).
RESULTS
Clinical
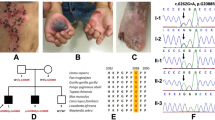
This large five generation family of Palestinian Arab origin of Muslim ancestry comprises 35 individuals, of which 6 are affected with a recessive form of epidermolysis bullosa (Fig. 1). Affected individuals presented at birth with blisters all over the body, including oral mucous membranes and genitalia (Fig. 2 a). At an older age, in addition to blisters, atrophy at healing sites was notable (Fig. 2 b). Teeth and fingernails were unaffected. Light microscopy of skin from affected individual V3 demonstrated dermal/epidermal separation with a mononuclear infiltration within the papillary dermis. Standard electron microscopy revealed lamina densa on the dermal side of the bullae, suggesting an intralamina lucida separation (not shown). Immunohistochemical staining for type IV collagen, type VII collagen and the γ2 chain of laminin 5 was equivalent to normal skin (not shown).

Pedigree of the five generation Palestinian family affected with GABEB. Affected members are shaded black. Individuals assessed in this study are marked with an asterisk (*).

Clinical appearance of affected family member (V.3) with GABEB. a, A blister in the toe finger presented at birth. b through d, Demarcation post blistering in foot, hand, and back at 3 years old.
Linkage analysis
Linkage analysis for markers on chromosomes 1 and 18 revealed no common disease allele for the affected individuals, thereby excluding linkage to the laminin 5 genes, LAMA3, LAMB3, and LAMC2. However, clear disease segregation was seen with markers on chromosome 10 demonstrating linkage to the type XVII collagen gene, COL17A1 (Fig. 3 a). In addition, as expected from the consanguineous nature of the family, the affected individuals demonstrated homozygosity by descent, suggesting that the disease was caused by a homozygous mutation in COL17A1.

Linkage and mutation analysis of COL17A1. a, COL17A1/10q linkage analysis and direct sequencing results for the family members tested. Disease allele is boxed. b, Detection of the homozygous splice site mutation IVS51+1G>A by direct nucleotide sequencing. Affected individuals are homozygous A at donor splice site +1, compared with heterozygous A/G carriers, and normal G homozygotes. The exon/intron boundary is marked.
Mutation findings
Direct sequencing of COL17A1 using patient DNA for the obligate carrier (IV.14) revealed a heterozygous G-to-A base substitution at the +1 position of the donor splice site in intron 51, termed IVS51+1G>A (Fig. 3 b). Subsequent sequencing of exon 51 demonstrated that all affected individuals were homozygous for the IVS51+1G>A mutation, that obligate carriers were heterozygous, and unaffected individuals were either heterozygous or homozygous for the normal allele (Fig. 3 a).
DISCUSSION
We demonstrate in this study a novel donor splice site mutation, IVS51+1G>A in COL17A1. Naturally occurring splice site mutations can be separated into three main groups. First, mutations within the 5′ or 3′ splice sites that can either lead to exon skipping, intron inclusion, or the use of a cryptic splice site, 21 resulting in either a truncated protein or a protein lacking a small region of the coding sequence. Second, mutations within an intron may lead to formation of a cryptic splice site that results in the inclusion of an aberrant exon within the transcript, and third, mutations within the branch point can result in the incorrect splicing out of the intron. However, the majority of splicing mutations occur within the invariant GT donor and AG acceptor splice sites, respectively. Mutations affecting any of these four bases will lead to a splicing defect, although the donor sequence has infrequently been found in vivo to be GC, which still permits correct splicing. 22 A similar mutation to the one in this study at the intron 51 donor splice site has been previously described, IVS51+1G>C. 23 Evaluation of RNA transcripts resulting of the latter mutation 23 had showed at least three aberrant COL17A1 transcripts. The first resulted in the in-frame skipping of the entire 147-bp exon 51, deleting 49 amino acids. The second also resulted in the in-frame skipping of exon 51, with the additional inclusion of 231 bp of intron 52 by activation of a cryptic splice site. This lead to premature truncation of the protein in codon 2 of the inserted intron 52 sequence. The third transcript is the result of the inclusion of a 56-bp portion of intron 51 due to cryptic splice site activation in intron 51, leading to a novel 77 amino acid carboxy terminus followed by premature truncation of the protein.
A homozygous donor splice site mutation in exon 52 of COL17A1 has also been described. 24 Transcript analysis demonstrated that the use of cryptic splice sites resulted in at least five different mutant transcripts, four of which resulted in in-frame deletions. 24 In-frame deleted transcripts may not be subjected to nonsense mediated mRNA decay, as is seen for COL17A1 transcripts that harbor nonsense mutations. 25 Thus, it can be postulated that the loss of some of the Gly-X-Y repeats within the carboxy terminal domain will destabilize the protein by preventing the formation of the collagen-like triple helices. Therefore, the mutation detected in the presented family is expected to result in aberrantly spliced COL17A1 gene transcript, leading to a defective protein. The severe phenotype detected in all 6 affected members of this family with no interfamilial variability support this assumption.
We assume that the mutation described in this study was introduced into the described family at least 5 generations ago by a founder couple comprising a carrier for the mutation. This couple (I.1 and I.2) and their two sons (II.1 and III.2) lived in Hebron. Three of their offspring (III.3, IV.1, and IV.13) moved to Lod, a city in Israel, highly populated with Arabs, and married unrelated local women. From there, intrafamilial marriages took place between the descendants of the settlers from Hebron. This increased the carrier pool and within 1 to 2 generations from the initial settlement in Lod homozygosity by descent occurred with disease presentation in 6 affected individuals, all of whom are cousins homozygous for the IVS51+1G>A mutation. As individuals III.1–2 and III.5–6 still live in Hebron, no DNA samples are available for mutation analysis due to boundary restrictions. Our assumption of mutation segregation in this family is based on the following facts: first, there are no reports of any affected patients with EB in the unrelated women's side in Lod, which include several tens of relatives in large families. Second, because III.4 (the wife of one of the brothers) is a normal homozygote, then her husband, the grandfather of 4 affected patients must have been a carrier. Therefore, his other 2 brothers from Hebron are also expected to be carriers and have transmitted the mutation from one of their parents who is suspected to be the founder. Because this autosomal recessive disease carries a high rate of morbidity with a considerable esthetic effect, the leader of the family (III.4) decided to seek genetic assistance for possible prenatal detection. Through a thorough teaching process, she now demands that unmarried individuals get spouses from outside of the community and encourage all pregnant women to get into the genetic system for evaluation.
References
Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol 2000; 42: 1051–1066.
Brown TA, Gil SG, Sybert VP, Lestringant GG, Tadini G, Caputo R Defective integrin α6β4 expression in the skin of patients with junctional epidermolysis bullosa and pyloric atresia. J Invest Dermatol 1996; 107: 384–391.
Hintner H, Wolff K Generalized atrophic benign epidermolysis bullosa. Arch Dermatol 1982; 118: 375–384.
Woo HJ, Lee JH, Kim SC, Kim CW, Kim TY Generalized atrophic benign epidermolysis bullosa-poor prognosis associated with chronic renal failure. Clin Exp Dermatol 2000; 25: 212–214.
Mazzanti C, Gobello T, Posteraro P, Paradisi M, Meneguzzi G, Chinni L 180-kDa bullous pemphigoid antigen defective generalized atrophic benign epidermolysis bullosa: report of four cases with an unusually mild phenotype. Br J Dermatol 1998; 138: 859–866.
Foldes C, Wallach D, Aubiniere E, Vignon-Pennamen MD, Cottenot F Generalized atrophic benign form of junctional epidermolysis bullosa. Dermatologica 1988; 176: 83–90.
Bircher AJ, Lang-Muritano M, Pfaltz M, Bruckner-Tuderman L Epidermolysis bullosa junctionalis progressiva in three siblings. Br J Dermatol 1993; 128: 429–435.
Swensson O, Christophers E Generalized atrophic benign epidermolysis bullosa in 2 siblings complicated by multiple squamous cell carcinomas. Arch Dermatol 1998; 134: 199–203.
Pohla-Gubo G, Lazarova Z, Giudice GJ, Liebert M, Grassegger A, Hintner H Diminished expression of the extracellular domain of bullous pemphigoid antigen 2 (BPAG2) in the epidermal basement membrane of patients with generalized atrophic benign epidermolysis bullosa. Exp Dermatol 1995; 4: 199–206.
Jonkman MF, de Jong MC, Heeres K, Pas HH, van der Meer JB, Owaribe K 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J Clin Invest 1995; 95: 1345–1352.
McGrath JA, Pulkkinen L, Christiano AM, Leigh IM, Eady RA, Uitto J Altered laminin 5 expression due to mutations in the gene encoding the β3 chain (LAMB3) in generalized atrophic benign epidermolysis bullosa. J Invest Dermatol 1995; 104: 467–474.
McGrath JA, Gatalica B, Christiano AM, Li K, Owaribe K, McMillan JR Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet 1995; 11: 83–86.
Anton-Lamprecht I Mutations in the laminin 5 LAMB3 gene in generalized atrophic benign epidermolysis bullosa. J Invest Dermatol 1995; 105: 856–857.
Nakano A, Chao S-C, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs non-Herlitz phenotypes. Hum Genet 2002; 110: 41–51.
Li KH, Sawamura D, Giudice GJ, Diaz LA, Mattei MG, Chu ML Genomic organization of collagenous domains and chromosomal assignment of human 180-kDa bullous pemphigoid antigen-2, a novel collagen of stratified squamous epithelium. J Biol Chem 1991; 266: 24064–24069.
Gatalica B, Pulkkinen L, Li K, Kuokkanen K, Ryynanen M, McGrath JA Cloning of the human type XVII collagen gene (COL17A1), and detection of novel mutations in generalized atrophic benign epidermolysis bullosa. Am J Human Genet 1997; 60: 352–365.
Giudice GJ, Emery DJ, Diaz LA Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol 1992; 99: 243–250.
Balding SD, Diaz LA, Giudice GJ A recombinant form of the human BP180 ectodomain forms a collagen-like homotrimeric complex. Biochemistry 1997; 36: 8821–8830.
Nishizawa Y, Uematsu J, Owaribe K HD4, a 180 kDa bullous pemphigoid antigen, is a major transmembrane glycoprotein of the hemidesmosome. J Biochem Tokyo 1993; 113: 493–501.
Masunaga T, Shimizu H, Yee C, Borradori L, Lazarova Z, Nishikawa T The extracellular domain of BPAG2 localizes to anchoring filaments and its carboxyl terminus extends to the lamina densa of normal human epidermal basement membrane. J Invest Dermatol 1997; 109: 200–206.
Berget SM Exon recognition in vertebrate splicing. J Biol Chem 1995; 270: 2411–2414.
Shapiro MB, Senapathy P RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987; 15: 7155–7174.
Pulkkinen L, Marinkovich MP, Tran HT, Lin L, Herron GS, Uitto J Compound heterozygosity for novel splice site mutations in the BPAG2/COL17A1 gene underlies generalized atrophic benign epidermolysis bullosa. J Invest Dermatol 1999; 113: 1114–1118.
van Leusden MR, Pas HH, Gedde-Dahl T, Sonnenberg A, Jonkman MF Truncated type XVII collagen expression in a patient with non-Herlitz junctional epidermolysis bullosa caused by a homozygous splice-site mutation. Laboratory Investigation 2001; 81: 887–894.
Huber A, Yee C, Darling TN, Yancey KB Comprehensive analysis of gene expression profiles in keratinocytes from patients with generalized atrophic benign epidermolysis bullosa. Exp Dermatol 2002; 11: 75–81.
Acknowledgements
We are very grateful to individual III.4, the leader of the family, for her devotion and dedication to this project. We also wish to thank the Royal Devon and Exeter Hospital.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Whittock, N., Sher, C., Gold, I. et al. A founder COL17A1 splice site mutation leading to generalized atrophic benign epidermolysis bullosa in an extended inbred Palestinian family from Israel. Genet Med 5, 435–439 (2003). https://doi.org/10.1097/01.GIM.0000096494.61125.D8
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000096494.61125.D8
