
Abstract
Expression quantitative trait loci (eQTL) studies have functionalized nucleic acid variants through the regulation of gene expression. Although most eQTL studies only examine the effects of single variants on transcription, a more complex process of variant–variant interaction (epistasis) may regulate transcription. Herein, we describe a tool called interaction QTL (iQTL) designed to efficiently detect epistatic interactions that regulate gene expression. To maximize biological relevance and minimize the computational and hypothesis testing burden, iQTL restricts interactions such that one variant is within a user-defined proximity of the transcript (cis-regulatory). We apply iQTL to a data set of 183 smallpox vaccine study participants with genome-wide association study and gene expression data from unstimulated samples and samples stimulated by inactivated vaccinia virus. While computing only 0.15% of possible interactions, we identify 11 probe sets whose expression is regulated through a variant–variant interaction. We highlight the functional epistatic interactions among apoptosis-related genes, DIABLO, TRAPPC4 and FADD, in the context of smallpox vaccination. We also use an integrative network approach to characterize these iQTL interactions in a posterior network of known prior functional interactions. iQTL is an efficient, open-source tool to analyze variant interactions in eQTL studies, providing better understanding of the function of epistasis in immune response and other complex phenotypes.
Similar content being viewed by others


Introduction
A recent emphasis on transcript-level variation has resulted in an increasing popularity of expression quantitative trait loci (eQTL) mapping studies. These studies examine the association between single-nucleotide polymorphisms (SNPs) and expression of transcripts. Although eQTL studies have begun to elucidate the variant-mediated nature of the transcriptome, a more complex process likely controls the mechanism of transcript regulation. Statistical epistasis, defined as variant–variant interactions that mediate complex phenotypes such as gene expression, likely contributes to the variation in observable traits. Given the high dimensionality and computational complexity of calculating interacting variants and transcript outcomes, epistatic models have had limited application to eQTL data. Herein, we describe a tool called interaction QTL (iQTL), designed to identify functional interactions between pairs of SNPs that regulate individual gene expression.
In eQTL terminology, cis-acting SNPs refer to variants that regulate a nearby transcript on the same chromosome. When variant regulation affects transcription on a different chromosome, the SNP is denoted as a trans-acting SNP. The recognition and identification of trans-acting eQTL SNPs are of particular interest as these variants affect downstream genes not implicated by traditional genome-wide association study (GWAS) methods.1 Although previous eQTL studies have rigorously identified single-variant regulation for thousands of transcripts, relatively few studies2, 3 have attempted to understand the effect of multiple SNPs interacting in concert (i.e., epistatically) to regulate gene expression. As multi-SNP transcript regulation cannot be detected in single-locus models,2 separate epistatic eQTL analyses must be performed to identify interactive effects.
Statistical epistasis, defined as the interaction between genes, is commonly estimated using a multiplicative term in a generalized linear model.4 Although presently represented in statistical models, the concept of an interaction was originally characterized when William Bateson coined the term ‘epistasis’ to explain the discrepancy between the prediction of segregation ratios in the outcome of a dihybrid cross.5 For the purposes of this manuscript, an appropriate general definition for a genetic interaction in the -omics era constitutes a statistically significant difference in a measurable phenotypic value conditioned on alleles from different loci in combination beyond their independent contributions.5
While epistatic interactions likely affect complex dichotomous phenotypes, there are advantages to characterizing interactions in quantitative traits, as they may provide a better resolution of the phenotypic effect. For example, interactions in quantitative trait loci in model organisms have demonstrated the biological importance of modeling these effects.6 In humans, the analysis of gene interactions has produced novel insights into not just gene expression but other quantitative traits such as cholesterol levels.7 The importance of characterizing interactions is apparent not just in characterizing the underlying biological processes that produce quantitative phenotypes but also in estimating the heritability of these traits.8
Although several existing packages for epistasis could be applied to discover epistatic eQTLs,3 no previous software package has been specifically designed to identify multi-SNP regulation of transcripts. For example, Becker et al.2 identified epistatic eQTLs in 210 HapMap individuals using PLINK’s epistasis command9 but immediately discarded nearly 99% of the computed pairwise interactions. The authors only examined instances of cis–trans-regulated expression, meaning exactly one of the two interacting SNPs had to fall in some proximity of the transcript, and they discarded instances of cis–cis and trans–trans interactions.2 Becker et al.2 acknowledged the difficulty in controlling for linkage disequilibrium in cis–cis interactions and multiple testing in trans–trans interactions to justify their exclusion. The iQTL tool in the current study implements these restrictions and minimizes the computational burden in terms of CPU time and disk storage.
To demonstrate the utility of iQTL, we applied the tool to an eQTL data set of vaccinia-stimulated and -unstimulated cells from a study of smallpox vaccines. The aim of this study was to discover genetic determinants of differential response of cells to the stimulation with vaccinia based on the genotype of variants in immune system genes. As previous studies have attempted to understand how variability of genetic information affects response to vaccines, we anticipated that the application of iQTL to this data set would better characterize the immune response using a higher-order regression model.10 Among the statistically significant epistatic eQTLs, we uncovered an enrichment of genes involved in apoptosis, a pathway that has been implicated in the immune response to influenza vaccine.11 In addition to identifying genetic determinants in the epistatic eQTL construct, we aimed to characterize the differences, if any, between cis- and trans-acting elements in an integrative network. We hypothesized that trans-acting elements would be more highly interactive in a systems biology framework, leading to a cascade of genetic effects that could produce phenotypic variance.
Results
A limiting factor of the application of epistatic models to quantitative trait loci studies is the tremendous computational burden. However, our implementation of iQTL restricts trans–trans interactions and substantially reduces the degree of the computational burden and affords users the ability to compute epistatic effects in their own data. In our application to the smallpox eQTL data, over 37.7 trillion interactions were computed by requiring SNPs to be characterized as cis–cis or cis–trans. This total represents <0.15% of possible pairwise interactions that other tools would normally compute from the SNPs and probe sets in the epistasis analysis. Additionally, the full output from iQTL for our analysis exceeded 2.45 terabytes. We note that the output from a similar analysis performed using PLINK’s epistasis would be on the order of thousands of terabytes. Thus, iQTL’s removal of trans–trans interactions from consideration via preprocessing saves computational burden, hypothesis burden and disk space on the order of 103, a necessary innovation to execute this analysis.
After computing all instances of cis–trans epistasis from over 17 290 microarray gene expression probe sets and 53 684 SNPs, 11 epistatic eQTL interactions (involving 19 genes and 20 unique SNPs) were significant at a Bonferroni-corrected threshold of 1.33 × 10−12 (Table 1). We note that the two effects of 207801_s_at are likely an artifact of linkage disequilibrium between rs2698042 and rs2730427 (r2=0.78) and not two unique epistatic effects. Although these 11 interactions were significant after correcting for multiple testing, we note that none of these interactions would be significant using a Bonferroni correction had all instances of trans–trans been computed (threshold: P=2 × 10−15). Thus, not only would this analysis under a full interaction model have been extremely prohibitive due to computational and hard disk burden but also the significant interactions discovered by iQTL would have been pruned under typical correction for multiple testing. We use a circular genome network (Figure 1) to provide an overview of the significant interactions discovered in this analysis and their relative positions on their respective chromosomes.

A circular genome network of significant epistasis eQTL effects produced by EINviz.42 The red lines connect the variants found to have a statistically significant interaction after correcting for multiple testing. These variants are then mapped to their respective genes and chromosomes in outer layers.
Using the 19 genes identified through the iQTL (Table 1), we characterize their effect in an integrative network approach. Using the Integrated Multi-species Prediction server (IMP), we inferred the connectivity of these 19 genes based on prior biological knowledge.12 The combined interaction network (Figure 2) includes genes identified in the iQTL analysis and distinguishes cis (circle) from trans (diamond) effects as well as interactions from the epistasis analysis (solid line), inferred from IMP (dashed line), or both (double line). Using this network, we tested our hypothesis that trans-acting elements are more highly connected. Indeed, the trans-acting genes had a significantly (P=0.0226) higher average degree (3.56) in the network than the average degree of cis-acting genes (2.25).

A network summary of significant epistasis eQTL effects. Genes in the green diamonds are trans effects and the blue circles represent cis effects. Solid lines represent significant iQTL interactions (P<1.33 × 10−12) and the dashed lines represent interactions inferred from IMP.12 The double barred line between TRAPPC4 and DIABLO represents an interaction produced by both IMP and iQTL.
To quantify the ability of iQTL to identify novel effects, we used the eQTL data to determine whether any epistatic model terms could be discovered using a univariant eQTL filter. Only 6 of the 21 unique variants had a nominal (uncorrected P<0.05) eQTL effect for the identified epistatic eQTL effects, including several trans effects (Table 1). Consequently, the interactions identified in the epistatic model would be removed by a univariate filter or even an additive regression model, which is consistent with a previous finding.2 We illustrate one of the significant epistatic eQTL effects by plotting the variation in the expression of a probe set mapping to SLC39A8 conditioned on the genotypes of the two SNPs that produced a significant epistatic interaction, rs3774937 and rs2832157 (Figure 3). While the average expression across most genotype groups has relatively low variance, the individuals with four minor alleles have significantly lower fold changes. This plot suggests that to observe the reduction in gene expression in response to vaccinia stimulation due to these SNPs, both SNPs must be present in an epistatic interaction as suggested in Table 1.

Boxplot of a significant epistasis eQTL effect for SLC39A8.The horizontal axis indicates the genotype combinations for the variants in epistasis, and the vertical axis shows the log2 fold change of the gene expression of stimulated cells over unstimulated cells for the 209267_s_at probe set (SLC39A8). (a) The boxplots for the given raw genotype combination. (b) The same information but the genotype encoding uses the product of the minor allele count (0, 1, 2) for rs3774937 multiplied by the minor allele count for rs2832157.
Discussion
While the framework needed to determine epistatic QTLs exists in other tools such as PLINK,9 iQTL provides the first dedicated option tailored to discovering the regulation of transcripts by multiple SNPs. By removing instances of trans–trans interactions and using parallel computations, iQTL increases the likelihood of biological relevance while greatly reducing computational and disk space burden by several orders of magnitude. Although it is generally accepted that statistical interactions or epistasis have some role in understanding the genetic basis of complex phenotypes, the biological interpretation of statistical SNP–SNP interactions has been difficult to ascertain.13 Some have hypothesized that the epistatic regulation of the transcriptome is an important intermediate step to understanding the role of epistasis in disease and other complex phenotypes.14 iQTL tests for these novel effects not included in single-locus eQTL models, thereby allowing for more complex models of variant-level regulation of transcription. We argue that using iQTL in eQTL studies will further elucidate the relationship between SNPs and the transcriptome.
Although considerable progress has been made using univariant models in genome-wide association and eQTL studies, we recently demonstrated the success of several network-based approaches for discovering novel immunological genetic correlates in systemic lupus erythematosus,15 influenza vaccine immune response16 and antibody response to the smallpox vaccine.17 Here, we computed interactions using iQTL and situated the significant interactions in an integrative network, thereby enabling a systems biology interpretation of the results. This approach using higher-order models to generate networks has previously been theorized to enhance the field of vaccinomics.18, 19 Our tool, coupled with network inference, provides a unique perspective on the role transcripts play in immune response and implicates the function of common variants in regulating response.
In our integrated network structure, we observed one interaction between DIABLO and TRAPPC4 that was inferred both by IMP and our epistatic interaction model. Interestingly, both genes participate in the stimulation of apoptosis by various cellular pathways. The trans-acting effect, DIABLO, has been shown to promote apoptosis by preventing several inhibitors of apoptosis proteins from binding to caspases.20 The partner from the epistatic model, TRAPPC4, has been shown to regulate apoptosis by interacting with extracellular signal-regulated kinase proteins.21 As IMP inferred a strong connection between these genes via strong coexpression in a large number of microarray studies, the synergistic effect of both genes promoting apoptosis could lead to more observable effects in complex phenotypes, including response to vaccinia stimulation. The results of our iQTL regression analysis implicate the role of epistasis between rs11216930 and rs550349 in leading to differential expression of TRAPPC4, which we hypothesize differentially affects apoptosis in response to the smallpox vaccine.
In addition to DIABLO and TRAPPC4 identified above, several genes identified in the epistatic analysis help regulate apoptosis, including FADD,22 ITK23 and REG3A.24 Although specific genetic correlates have not been previously identified that link differential smallpox vaccine response to apoptosis, this cellular pathway has been implicated in immune response for other vaccines.11 Previous work has demonstrated that apoptotic cellular death after viral infection can trigger strong innate and adaptive immune responses.25 Similarly, carefully controlled cell death shapes both the naïve lymphocyte pool26 and optimal clonal expansion of antigen-specific T and B cells.27 Specifically, apoptotic but not necrotic cellular vaccinations have been shown to induce potent immune response in vivo.28 Although the role of apoptosis has been implicated in immune response, univariant models applied to our smallpox genotyping or microarray data set failed to identify these effects. Vaccinia virus and other poxviruses encode multiple inhibitors of apoptosis, presumably as a sophisticated mechanism of immune evasion and to enhance viral replication. Additionally, poxviruses such as vaccinia preferentially infect professional antigen-presenting cells such as macrophages and dendritic cells.29, 30 Differential activity of the apoptotic pathways in these infected cells may markedly alter the amount and type of viral antigens available for presentation to the adaptive immune system. The utilization of a higher-order interaction model discovered these genetic effects linked to apoptosis.
Although a limitation of the present application of iQTL is the small sample size of the eQTL data set, the increasing affordability of expression and genotyping data will lead to larger sample sizes in future analyses. Consequently, computationally efficient tools are needed to infer significant results from large data sets, particularly epistatic effects that require higher-dimensional models. Computational efficiency becomes even more critical when the genotyped individuals represent a diverse population, often requiring covariates from principal component analysis to adjust for potential confounding via population stratification. While the additional network inference supports the biological activity of the results, further applications of iQTL in larger sample sizes are required to validate this finding. Although further characterization of these genes is required to better understand their role in personalized vaccine response, iQTL is the first tool that enables the discovery of these effects on a reasonable computational scale.
Materials and methods
iQTL implementation
iQTL is written and implemented in C++ through modification of the PLINK source code.9 This design retains much of the functionality of PLINK while creating our new commands that match the user-friendly command-line interface provided by this popular tool.9 Instead of computing interactions sequentially, iQTL is implemented in the OpenMP (Open Multi-Processing) framework to enable the division of computational burden across all available cores on a machine or server.2, 31 By implementing iQTL in C++, excluding unnecessary computations, and parallelizing the interaction computations, our tool incorporates key features necessary for efficient bioinformatics software.14, 16
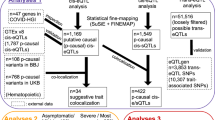
The key innovation that iQTL provides is the elimination of unnecessary pairwise SNP computation, as only instances of cis–cis and cis–trans are computed. When a user executes the iQTL command, SNPs are categorized as either cis or trans for each transcript. A user can reduce the classified cis SNPs by using the radius flag to define a region around the transcript; the default is set at 1000 kilobases (kb). iQTL then computes the standard eQTL association for each SNP with transcript within this region using the linear regression model:

where the response variable Expression is a measure of gene expression for the ith person, β0 and β1 are the intercept and slope, respectively, SNP1 takes on values of (0, 1, 2) according to the number of minor alleles and ɛi are random errors assumed to independently distributed Gaussian random variables. These eQTL associations are calculated automatically and can be used to quickly interpret results from the interaction QTL model. For example, these univariant models can be used to interpret whether the interaction determined is the composition of two significant eQTL effects or a novel effect altogether. For two SNPs in the iQTL model, we use the linear regression equation:

where model terms are analogous to the eQTL model. The significance of the interaction between the two SNPs is determined by the test statistic associated with the β3 coefficient in the interaction model. We note that using regression models easily enables the use of covariates when performing the association analyses. A complete user’s guide and additional details on the implementation and execution are available on our website.
Smallpox eQTL data set
For gene expression profiling, 200 subjects were selected based on immune extremes (both high and low) expression of humoral (neutralizing antibody) and cellular (INF-γ Elispot) responses to primary smallpox vaccination among 1076 healthy, eligible individuals. These subjects ranged from ages 18 to 40 years, and these demographic and immune variables have been previously described in depth.32, 33 The participants were recruited as participants in a smallpox immunization program at the Naval Health Research Center in San Diego, CA, and the Department of Health and Human Services civilian healthcare worker smallpox immunization program at the Mayo Clinic in Rochester, MN.33, 34 All study subjects were immunized with a single dose of Dryvax smallpox vaccine (Wyeth Laboratories Inc., Marietta, PA, USA) within 4 years before recruitment, and had a documented vaccine vesicular ‘take’, or development of a pustule, at the vaccination site.
Four gender-specific quadrants were defined based on gender-specific medians to define high (above the median) and low (below the median) responses as described previously.32 For each of the humoral and cellular responses, the squared difference from the median was calculated per subject and then scaled to range from 0 to 1 for all 1076 subjects. For each subject, the product of the humoral and cellular scaled, and then squared deviations were computed. Twenty-five males and 25 females having the largest of these values in each quadrant were chosen for subsequent microarray analysis.
To improve cell viability, peripheral blood mononuclear cells were rested overnight with 50 IU/ml of interleukin-2, as described previously.35 While one half of the cells for each patient were left unstimulated, we stimulated half of the cells with inactivated vaccinia virus at a multiplicity of infection of 0.5 plaque-forming units per cell for 18 h as described previously.36 We stabilized the cells using RNAprotect cell reagent (Qiagen, Valencia, CA, USA) and extracted total RNA using RNeasy Plus Mini Kit (Qiagen). We assessed RNA quantity and quality by Nanodrop spectrophotometry (Thermo Fisher Scientific, Wilmington, DE, USA) and Agilent 2100 Bioanalyzer Chip Kit Analysis (Agilent, Palo Alto, CA, USA). All microarray experiments were performed at the Mayo Advanced Genomics Technology Center Microarray Shared Resource core facility using hybridization to Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix Corp., Santa Clara, CA, USA) following a standardized operating procedure and the manufacturer’s specifications.
Each of the 1076 individuals was genotyped as reported previously.37 DNA was extracted from whole blood or blood clots using the Gentra Puregene Blood Kit (Gentra Systems Inc., Minneapolis, MN, USA) and quantified by Picogreen (Molecular Probes, Carlsbad, CA, USA). High-density SNP analysis was performed using the Infinium BeadChip Array (Illumina, San Diego, CA, USA) described as previously in depth.37 Initial QC steps were used to remove suspect SNPs such as those with poor clustering, monomorphic SNPs or those that failed Hardy–Weinberg equillibrium.37 Seventy-one subjects with low-quality genotyping (call rates <95%) were also excluded.37 The intersection of the samples who passed genotyping quality control and who had the stimulated and unstimulated microarray analysis performed resulted in an eQTL sample size of 183 (88 males and 95 females). The Institutional Review Boards of the Mayo Clinic and NHRC approved the study and written informed consent was obtained from each subject.
iQTL smallpox analysis
Although iQTL significantly reduces the computational burden by restricting the regression model to cis–cis and cis–trans interactions, the three-dimensional (number of SNPs2 × number of probe sets) nature of the models necessitated further filtering to create a computationally feasible approach. Using prior knowledge of genes related to the immune system, we restricted expression probe sets that mapped to one of 4815 genes from the ImmPort Database.38 The list of immunologically related genes in ImmPort is a collection of ~6000 human genes, which was formed with the aim of retrieving all genes that have immune system-related functions.38 Although the list was generated using automatic searches of EntrezGene and Gene Ontology records using immunology-related keywords, the final list was manually curated by immunology experts examining various literature sources, leading to a final collection of 4815 genes contained in InnateDB.38 Seventeen thousand two hundred and ninety expression probe sets from the microarray platform were successfully mapped onto these 4815 genes and were selected for further analysis by iQTL. While the software automatically removes the instances of trans–trans interactions, other filtering based on prior biological knowledge could be used. In this study, the log2 fold change of the stimulated expression over the unstimulated expression was the quantitative trait of interest.
To reduce the dimension of the SNP data, two additional measures were taken in addition to the standard quality control described previously.37 First, only variants whose allele frequencies ranged between 0.2 and 0.8 were selected. While the standard common variant threshold is 5%, this more stringent requirement yields greater representation in genotype groups with several minor alleles. For example, two individuals had all four minor alleles in Figure 3, whereas one would not expect any individual to have four copies of minor alleles occurring at 5% in the current sample size. Second, variants had to be positioned within 500 kb of one or more of the 4815 ImmPort genes to be selected for analysis. This is similar to what other studies have used in eQTL analyses,39 and represents a more conservative genomic mapping than other eQTL studies that use 1 Mb.2 These additional filtering processes left 53 684 SNPs for analysis in iQTL. Figure 4 shows a graphical overview of the regression model used for a cis–trans interaction in iQTL.

A schematic summary of the regression model used for a cis–trans interaction. A log2 fold change of the expression of a probe set for stimulated over unstimulated mRNA was regressed against the standard epistasis linear model for one cis (within 500 kb of transcript) and one trans SNP. Chromosome A contains the transcript and cis SNP, whereas chromosome B contains the trans SNP.
Secondary network analyses
After computing the significant epistatic interactions displayed in Table 1, we used IMP in an integrative network approach to better understand our implicated iQTL interactions.12 Although the significant interactions themselves provide novel insight into the variability of the genetic response of vaccination, additional insights can be inferred by analyzing effects of variants (nodes) in a network topology.40 Specifically, we recently demonstrated the efficacy of using IMP to characterize results from higher-order models applied to eQTL data sets.41 Thus, to infer the connections between genes identified in the iQTL analysis, edges were determined using IMP with a 0.1 posterior confidence threshold. The centrality or connectedness in the cis and trans genes was assessed by comparing the distribution of network degree from the connected graph of 17 nodes, and the significance was determined using the unpaired Student’s T-test.
Code availability
The iQTL software for linux and a tutorial are available at http://insilico.utulsa.edu/index.php/iqtl.
References
Fehrmann RS, Jansen RC, Veldink JH, Westra HJ, Arends D, Bonder MJ et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet 2011; 7: e1002197.
Becker J, Wendland JR, Haenisch B, Nothen MM, Schumacher J . A systematic eQTL study of cis-trans epistasis in 210 HapMap individuals. Eur J Hum Genet 2012; 20: 97–101.
Kapur K, Schupbach T, Xenarios I, Kutalik Z, Bergmann S . Comparison of strategies to detect epistasis from eQTL data. PLoS One 2011; 6: e28415.
Cordell HJ . Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet 2002; 11: 2463–2468.
Phillips PC . Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 2008; 9: 855–867.
Liti G, Louis EJ . Advances in quantitative trait analysis in yeast. PLoS Genet 2012; 8: e1002912.
Ma L, Clark AG, Keinan A . Gene-based testing of interactions in association studies of quantitative traits. PLoS Genet 2013; 9: e1003321.
Zuk O, Hechter E, Sunyaev SR, Lander ES . The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA 2012; 109: 1193–1198.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Oberg AL, McKinney BA, Schaid DJ, Pankratz VS, Kennedy RB, Poland GA . Lessons learned in the analysis of high-dimensional data in vaccinomics. Vaccine 2015; 33: 5262–5270.
Furman D, Jojic V, Kidd B, Shen-Orr S, Price J, Jarrell J et al. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol Syst Biol 2013; 9: 659.
Wong AK, Park CY, Greene CS, Bongo LA, Guan Y, Troyanskaya OG . IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res 2012; 40: W484–W490.
Cordell HJ . Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet 2009; 10: 392–404.
Huang Y, Wuchty S, Przytycka TM . eQTL epistasis—challenges and computational approaches. Front Genet 2013; 4: 51.
Davis NA, Lareau CA, White BC, Pandey A, Wiley G, Montgomery CG et al. Encore: Genetic Association Interaction Network centrality pipeline and application to SLE exome data. Genet Epidemiol 2013; 37: 614–621.
Lareau CA, White BC, Oberg AL, McKinney BA . Differential co-expression network centrality and machine learning feature selection for identifying susceptibility hubs in networks with scale-free structure. BioData Min 2015; 8: 5.
Davis NA, Crowe Jr JE, Pajewski NM, McKinney BA . Surfing a genetic association interaction network to identify modulators of antibody response to smallpox vaccine. Genes Immun 2010; 11: 630–636.
Poland GA, Kennedy RB, McKinney BA, Ovsyannikova IG, Lambert ND, Jacobson RM et al. Vaccinomics, adversomics, and the immune response network theory: individualized vaccinology in the 21st century. Semin Immunol 2013; 25: 89–103.
McKinney BA, White BC, Grill DE, Li PW, Kennedy RB, Poland GA et al. ReliefSeq: a gene-wise adaptive-K nearest-neighbor feature selection tool for finding gene–gene interactions and main effects in mRNA-Seq gene expression data. PLoS One 2013; 8: e81527.
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43–53.
Zhao SL, Hong J, Xie ZQ, Tang JT, Su WY, Du W et al. TRAPPC4-ERK2 interaction activates ERK1/2, modulates its nuclear localization and regulates proliferation and apoptosis of colorectal cancer cells. PLoS One 2011; 6: e23262.
Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM . FADD a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81: 505–512.
Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M . The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 2001; 23: 436–446.
Choi B, Suh Y, Kim WH, Christa L, Park J, Bae CD . Downregulation of regenerating islet-derived 3 alpha (REG3A) in primary human gastric adenocarcinomas. Exp Mol Med 2007; 39: 796–804.
Restifo NP . Building better vaccines: how apoptotic cell death can induce inflammation and activate innate and adaptive immunity. Curr Opin Immunol 2000; 12: 597–603.
Smith KG, Light A, O’Reilly LA, Ang SM, Strasser A, Tarlinton D . Bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J Exp Med 2000; 191: 475–484.
Zhou T, Edwards CK, Mountz JD . Prevention of age-related T cell apoptosis defect in CD2-fas-transgenic mice. J Exp Med 1995; 182: 129–137.
Scheffer SR, Nave H, Korangy F, Schlote K, Pabst R, Jaffee EM et al. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer 2003; 103: 205–211.
Lehman MH, Price PJ, Brandmuller C, Sutter G . Modified Vaccinia virus Ankara but not vaccinia virus induces chemokine expression in cells of the monocyte/macrophage lineage. Virol J 2015; 12: 12–21.
Liu L, Chavan R, Feinberg MB . Dendritic cells are preferentially targeted among hematolymphocytes by Modified Vaccinia Virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol 2008; 9: 15.
OpenMP. Application Program Interface, 2008. Available at: http://www.openmp.org/mp-documents/spec30.pdf accesed 22 March 2016.
Haralambieva IH, Oberg AL, Dhiman N, Ovsyannikova IG, Kennedy RB, Grill DE et al. High-dimensional gene expression profiling studies in high and low responders to primary smallpox vaccination. J Infect Dis 2012; 206: 1512–1520.
Kennedy RB, Ovsyannikova IG, Pankratz VS, Vierkant RA, Jacobson RM, Ryan MA et al. Gender effects on humoral immune responses to smallpox vaccine. Vaccine 2009; 27: 3319–3323.
Haralambieva IH, Ovsyannikova IG, Dhiman N, Kennedy RB, O’Byrne M, Pankratz VS et al. Common SNPs/haplotypes in IL18R1 and IL18 genes are associated with variations in humoral immunity to smallpox vaccination in Caucasians and African Americans. J Infect Dis 2011; 204: 433–441.
Umlauf BJ, Ovsyannikova IG, Haralambieva IH, Kennedy RB, Vierkant RA, Pankratz VS et al. Correlations between vaccinia-specific immune responses within a cohort of armed forces members. Viral Immunol 2011; 24: 415–420.
Oberg AL, Dhiman N, Grill DE, Ryan JE, Kennedy RB, Poland GA . Optimizing high dimensional gene expression studies for immune response following smallpox vaccination using Taqman(R) low density immune arrays. J Immunol Methods 2011; 366: 69–78.
Kennedy RB, Ovsyannikova IG, Pankratz VS, Haralambieva IH, Vierkant RA, Jacobson RM et al. Genome-wide genetic associations with IFNgamma response to smallpox vaccine. Hum Genet 2012; 131: 1433–1451.
Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res 2014; 58: 234–239.
Turner SD, Bush WS . Multivariate analysis of regulatory SNPs: empowering personal genomics by considering cis-epistasis and heterogeneity. Pac Symp Biocomput 2011: 276–287.
Lareau CA, McKinney BA . Network theory for data-driven epistasis networks. Methods Mol Biol 2015; 1253: 285–300.
Lareau CA, White BC, Montgomery C, McKinney BA . dcVar: a method for identifying common variants that modulate differential correlation structures in gene expression data. Front Genet 2015; 6: 312.
Wu Y, Zhu X, Chen J, Zhang X . EINVis: a visualization tool for analyzing and exploring genetic interactions in large-scale association studies. Genet Epidemiol 2013; 37: 675–685.
Acknowledgements
Research reported in this publication was supported by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health, Department of Health and Human Services under award number U01IOFAI89859. This project was funded by federal funds from the National Institute of Allergies and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN266200400025C (N01AI40065).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Poland is the chair of a Safety Evaluation Committee for novel investigational vaccine trials being conducted by Merck Research Laboratories. Dr Poland offers consultative advice on vaccine development to Merck & Co. Inc., CSL Biotherapies, Avianax, Dynavax, Novartis Vaccines and Therapeutics, Emergent Biosolutions, Adjuvance and Microdermis. Dr Poland holds two patents related to vaccinia and measles peptide research. These research activities have been reviewed by the Mayo Clinic Conflict of Interest Review Board and are conducted in compliance with Mayo Clinic Conflict of Interest policies.
Rights and permissions
About this article

Cite this article
Lareau, C., White, B., Oberg, A. et al. An interaction quantitative trait loci tool implicates epistatic functional variants in an apoptosis pathway in smallpox vaccine eQTL data. Genes Immun 17, 244–250 (2016). https://doi.org/10.1038/gene.2016.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gene.2016.15
