
Abstract
Purpose
The post-LASIK exacerbation of corneal dystrophy, otherwise asymptomatic, is almost exclusively associated with the TGFBI gene mutations at codon 124 in exon 4 and codon 555 in exon 12. It is our intention to demonstrate that the pre-operative genetic screening for TGFBI mutations should be mandatory for refractive surgery candidates.
Patients and Methods
In this study, we reviewed the proband’s post-LASIK slit-lamp and in vivo confocal microscopy images and genetic testing results, and performed genetic testing on eleven additional members of the family to investigate the penetrance of corneal dystrophy in asymptomatic members who carry the mutation.
Results
The proband demonstrated a post-LASIK exacerbation of Granular Corneal Dystrophy type 2 (GCD2), identified as a TGFBI R124H mutation. Three of the 11 family members tested positive for the same R124H mutation as the proband.
Conclusion
The lesson learned from this case is that the genetic screening of TGFBI mutations must be incorporated into the pre-operative screening procedures to prevent exacerbation and recurrence, which eventually could lead to the need for a corneal transplant.
Similar content being viewed by others
Introduction
As the first report of autosomal-dominant Granular Corneal Dystrophy type 2 (GCD2) in individuals from Avellino, Italy1 in 1988, many cases of post-laser surgery exacerbation have been reported worldwide.1, 2, 3, 4, 5, 6, 7 Inherited corneal dystrophy exacerbation is characterized by bilateral opacity in anterior corneal stroma leading to a severe decrease of the best corrected visual acuity (BCVA) and ultimately to surgical treatment.8, 9, 10
The Avellino Universal Test examines the five most common TGFBI corneal dystrophies, each triggered by different mutations in exons 4 and 12 of the TGFBI gene, located on chromosome 5q31.1. Purified DNA is extracted from oral epithelial cells collected by buccal swabs and the genotype of the LASIK candidate obtained by amplifying the targeted DNA point mutations (Table 1).
Herein, we report the results of ophthalmic and genetic examination of an individual with post-LASIK GCD2 and his family from Jiangsu province, China.
Case series
Case 1
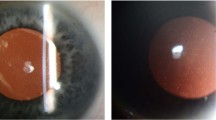
The proband is a 29-year-old Chinese male who underwent bilateral LASIK surgery in 2006. The post-operative uncorrected visual acuity (UCVA) in both eyes was 20/20. He had an uneventful post-operative course and did not have regular follow-up examinations after the surgery. In July 2016, 10 years after LASIK surgery, he was referred to Shanghai First People's Hospital for evaluation of dryness, foreign body sensation, and mildly decreased vision acuity in both eyes. The findings were opacities in both of his corneas. Slip-lamp examination was conducted (Figure 1a and b). His UCVA was 16/20 in the right eye and 12/20 in the left eye. The manifest refraction was −1.00 diopters cylinder × 30 oculus dexter and −0.5 oculus sinister, yielding a BCVA of 20/20 in the right eye and 16/20 in the left eye. Subsequent examination by in vivo confocal microscopy (IVCM) revealed a large quantity of dense white deposits of various shapes and sizes which presented as hyper-reflective extracellular structures located mainly in the anterior stroma (Figure 1c and d).

Slit-Lamp (Nikon Corporation, Tokyo, Japan) photographs of the proband revealed dense, fine, white granules in the central corneas of both eyes, located at the interface between the flap and stromal bed on the right (a) and left (b) eyes. In vivo confocal microscopy (IVCM) (Rostock Cornea Module of Retina Tomograph (HRT/RCM); Heidelberg Engineering GmbH, Heidelberg, Germany) images of the right (c) and left (d) anterior stroma with hyper-reflective extracellular deposits. In vivo confocal microscopy (IVCM) images of the right (e) and left (f) anterior stroma with hyper-reflected extracellular deposits observed in the corneas of the proband’s sister.
To uncover the cause of these deposits, the proband underwent genetic testing with the Universal Test (Avellino Labs China, Shanghai, China) which showed that he harbored a heterozygous R124H GCD2 mutation but not the R124C LCD1, R124L RBCD, R555W GCD1 or R555Q TBCD mutations (Table 1).
Case 2
The proband’s older sister is a 38-year-old female without any history of ocular surgery, eye complaints, or decreased vision. Following DNA testing, which revealed a heterozygous result for the R124H mutation, she was evaluated by IVCM which demonstrated similar hyper-reflective extracellular opacities at the anterior stroma with fewer, lower density deposits (Figure 1e and f).
Case 3
GCD2 is an autosomal-dominant disorder caused by the R124H TGFBI mutation. A study of 10 family members, in addition to the proband and his sister, was conducted and a family tree with test results was constructed (Figure 2d). The proband’s mother and his 15-year-old nephew (his sister’s son) also tested positive. Slit-lamp examination and IVCM were not conducted on these individuals due to limited access to a suitably equipped medical facility. Testing showed that the proband’s 7-year-old son had not inherited the mutation. The mutation status of the proband’s grandparents was unknown. Since the proband’s aunt tested negative and the medical history of the deceased uncle is unknown, it is unclear whether the proband’s mother inherited the mutation or it arose de novo.

The amplification plot displays normalized dye fluorescence (ΔRn) as a function of cycle number. The magenta slope in the amplification plot represents the normal allele (allele 1) and the blue slope represents the mutant allele (allele 2). (a) This plot shows the Proband’s sample of heterozygous mutation (blue slope) being amplified, which indicates the presence of mutation on allele 2. (b and c) amplification plots of GCD2 normal genotype and GCD2 homozygous mutation genotype for comparison purposes. (d) Family tree with test result of Heterozygous mutation plot. It is unknown whether the mutation was passed down from the proband’s grandmother or grandfather. From the family tree, it is clear that the proband’s mother carries the mutation, either inherited from the grandparents or caused by a de novo mutation. The proband’s mother did pass down the mutation to both of her children. The proband did not pass the mutation to his son. However, his 15-year-old nephew inherited the mutation from the proband’s sister.
Discussion
Corneal dystrophies contraindicate refractive surgery due to the likelihood of recurrence and exacerbation. As they may be difficult to determine by family history and clinical examination alone, a genetic test to detect TGFBI mutations should be incorporated into standard practice as one of the prescreening tools for refractive surgeries. In Case 1, the patient displayed no clinical symptoms and passed the LASIK surgery prescreening examination. Ten years after surgery, he developed symptoms that affected his vision. A genetic test identified that most likely the cause of the stromal deposits was the TGFBI R124H heterozygous mutation. The proband’s 15-year-old nephew inherited the mutation from proband’s sister, who tested positive for the mutation without symptoms; however, the examination conducted after the testing revealed that she too had corneal stromal deposits. These two cases demonstrate that a lack of clinical signs does not mean the absence of corneal dystrophy-causing mutations. It is justified to test the asymptomatic refractive surgery candidates to rule out disease-causing mutations.
Corneal dystrophy is a disease with a low prevalence (US: 1:1115,11 Korea: 1:870,12 China 1:4167) and debilitating outcome, ultimately resulting in corneal transplant as a treatment. The recurrent nature of the disease can result in multiple corneal transplants. Therefore, prevention and prescreening with a genetic test to detect the mutations, in addition to a thorough clinical examination is key.

References
Lee WB, Himmel KS, Hamilton SM, Zhao XC, Yee RW, Kang SJ et al. Excimer laser exacerbation of Avellino corneal dystrophy. J Cataract Refract Surgery 2007; 33 (1): 133–138.
Aldave AJ, Sonmez B, Forstot SL, Rayner SA, Yellore VS, Glasgow BJ . A clinical and histopathologic examination of accelerated TGFBIp deposition after LASIK in combined granular-lattice corneal dystrophy. Am J Ophthalmol 2007; 143: 416–419.
Banning CS, Kim WC, Randleman JB, Kim EK, Stulting RD . Exacerbation of Avellino corneal dystrophy after LASIK in North America. Cornea 2006; 25: 482–484.
Gruenauer-Kloevekorn C, Braeutigam S, Froster UG, Duncker GI . Surgical outcome after phototherapeutic keratectomy in patients with TGFBI-linked corneal dystrophies in relation to molecular genetic findings. Graefes Arch Clin Exp Ophthalmol 2009; 247: 93–99.
Jun RM, Tchah H, Kim TI, Stulting RD, Jung SE, Seo KY et al. Avellino corneal dystrophy after LASIK. Ophthalmology 2004; 111: 463–468.
Zeng L, Zhao J, Chen Y, Zhao F, Li M, Chao-Shern C et al. TGFBI gene mutation analysis of clinically diagnosed granular corneal dystrophy patients prior to PTK: a pilot study from Eastern China. Sci Rep 2017; 7: 596.
Song YZ, Sun MS, Qiu LM, Chen YG, Aldave AJ, Zhang FJ et al. The prevalence of TGFBI corneal dystrophies in Chinese refractive surgery candidates: primary analysis of 2068 cases in a multicenter study in China. J Cataract Refract Surgery. in press.
Ellies P, Bejjani RA, Bourges JL, Boelle PY, Renard G, Dighiero PL . Phototherapeutic keratectomy for BIGH3-linked corneal dystrophy recurring after penetrating keratoplasty. Ophthalmology 2003; 110: 1119–1125.
Inoue T, Watanabe H, Yamamoto S, Maeda N, Inoue Y, Shimomura Y et al. Recurrence of corneal dystrophy resulting from an R124H Big-h3 mutation after phototherapeutic keratectomy. Cornea 2002; 21: 570–573.
Szentmáry N, Seitz B, Langenbucher A, Schlötzer-Schrehardt U, Hofmann-Rummelt C, Naumann GO . Histologic and ultrastructural changes in corneas with granular and macular dystrophy after excimer laser phototherapeutic keratectomy. Cornea 2006; 25: 257–263.
Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A . Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci 2011; 52 (9): 6959–6963.
Lee JH, Cristol SM, Kim WC, Chung ES, Tchah H, Kim MS et al. Prevalence of granular corneal dystrophy type 2 (Avellino corneal dystrophy) in the Korean population. Ophthal Epidemiol 2010; 17 (3): 160–165.
Acknowledgements
We are thankful for all the unconditional support provided by Mr Gene Lee and the Avellino Labs China staff. In addition, we thank the support from the Department of Ophthalmology, Shanghai First People's Hospital, Shanghai Jiaotong University School of Medicine. Finally, we thank the family members who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
John Marshall is a consultant to Avellino Lab USA, Inc., Frost Professor at Institute of Ophthalmology, University College of London, London, UK. Tara Moore is a consultant to Avellino Lab USA, Inc., Director of the Biomedical Sciences Research Institute and Professor of Personalized Medicine at University of Ulster, Coleraine, Northern Ireland, UK. M Andrew Nesbit is a Senior Lecturer at University of Ulster, Coleraine, Northern Ireland, UK. Rao Me is an Ophthalmologist and BL Ke is Professor of Ophthalmology at Shanghai First People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China. Larry DeDionisio is an employee of Avellino Lab USA, Inc., Menlo Park, CA, USA. Connie Chao-Shern is a PhD student at University of Ulster, Coleraine, Northern Ireland, UK and an employee of Avellino Lab USA, Inc., Menlo Park, CA, USA. The authors declare no conflict of interests. The authors alone are responsible for the content and writing of this article.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chao-Shern, C., Me, R., DeDionisio, L. et al. Post-LASIK exacerbation of granular corneal dystrophy type 2 in members of a chinese family. Eye 32, 39–43 (2018). https://doi.org/10.1038/eye.2017.265
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2017.265
