
Abstract
Purpose
To perform mutational screening of the visual system homeobox gene 1 (VSX1; MIM#605020) in patients with sporadic and familial keratoconus (MIM#148300) in a European population and, for the first time, report the mutational analysis of the two newly identified VSX1exons.
Methods
VSX1sequence variants in patients with keratoconus were evaluated by direct sequencing of the entire coding region, including two novel exons. In familial keratoconus cases, segregation of potentially pathogenic VSX1variants was assessed to determine pathogenicity. Transcript analysis was carried out on splice site and synonymous sequence variants not detected in controls.
Results
A total of 66 unrelated patients with keratoconus from the European population (27 with familial keratoconus; 39 with sporadic keratoconus) were analysed for VSX1mutations. Four sequence variants were not observed in 100 healthy control individuals: c.432C>G (p.D144E), c.479G>A (p.G160D), c.789C>T (p.S263S), and an intronic change c.844-13T>A (numbered with respect to NM_014588). Segregation was not detected for p.D144E and c.844-13T>A. The change in p.G160D was observedin two patients with sporadic keratoconus. Although predicted to alter VSX1splicing, p.S263S had no effect on transcript processing. Four known SNPs were detected and the following polymorphic variants were observed in keratoconus patients and controls: c.711T>A (NM_199425; p.P237P), c.844-5_-6insT (NM_014588), c.*28G>T (DQ854811/DQ854812), and c.*50G>A (DQ854809/DQ854810).
Conclusions
VSX1has a minor role in keratoconus pathogenesis. The pathogenicity of p.G160D remains controversial and this change may represent a rare polymorphism or genetic modifier. Further evidence is provided that the previously reported variant, p.D144E, is a polymorphism.
Similar content being viewed by others

Introduction
Keratoconus (KCTN; MIM#148300) is a bilateral, non-inflammatory progressive corneal ectasia, manifesting as progressive myopia and irregular astigmatism, and is a major indicator for corneal transplantation in the Western world.1, 2 The estimated prevalence of keratoconus in the general population is ∼50–230 of 100 000,3 depending on the clinical detection method used and the diagnostic criteria applied. Keratoconus is believed to be inherited autosomally because of familial occurrence,4 with a higher concordance rate of the trait in monozygotic twins than in dizygotic twins,5 and its prevalence in first-degree relatives is 15–67 times higher than that of the general population.6 Most studies describe autosomal dominant inheritance, with incomplete penetrance or variable expressivity.7, 8, 9
The introduction of computerised videokeratoscopy or corneal topography has provided objective indices improving diagnostic accuracy and the ability to detect subclinical or ‘forme fustre’ keratoconus.10, 11, 12, 13 Mild corneal topographic anomalies have been reported among relatives of keratoconus patients14, 15 and may allow the detection of low expressivity forms of keratoconus, expanding pedigrees for mapping studies.14, 16, 17 Although most cases have been thought to be sporadic, the ability to detect subclinical cases with corneal topography has caused a revision of familial incidence by 6–8%.18
The genetic basis of keratoconus allows the application of linkage mapping and mutational analysis to elucidate its molecular basis and pathogenesis.19, 20, 21 Mapping studies have identified a number of loci for autosomal dominantly inherited keratoconus: 20p11-q11 (KTCN1; MIM#148300),22, 23 16q22.3-q23.1 (KTCN2; MIM#608932),9 3p14-q13 (KTCN3; MIM#608586),17 2p24 (KTCN4; MIM#609271),16 15q22.32-24,24, 25 and 5q14.3-q21.1.26 Other potential loci have been reported,27, 28 with likely further genetic heterogeneity.
Héon et al22 mapped a major gene for posterior polymorphous corneal dystrophy-1 (PPCD1; MIM#122000) to chromosome 20p11-q11, and subsequently identified mutations in the visual system homeobox gene 1 (VSX1; MIM#605020) in PPCD1 and keratoconus (KTCN1; MIM#148300).23 There has been debate in literature with regard to the significance of VSX1 in keratoconus development, with some authors questioning the role of VSX1 in keratoconus.29, 30 VSX1 was originally characterised as a 5-exon gene, spanning 6.7 kb of genomic DNA on chr20 with two major transcripts, 1 and 2 (NM_014588 and NM_199425), encoding protein isoform A (NP_199425.1) and B (NP_955457), respectively.31, 32 Recently, two novel exons downstream of the original VSX1 gene sequence were identified, which produce four additional previously unknown VSX1 transcripts.33 VSX1 is now known to span 10.65 kb of genomic DNA and consists of seven exons producing a total of six transcripts (Genbank NM_014588 also called DQ854807, NM_199425 also called DQ854808, DQ854809, DQ854810, DQ854811, and DQ854812).33 A further elucidation of the VSX1 transcript structure with two novel exons allows mutational analysis of the complete gene sequence for the first time and may explain the relatively low frequency of the pathogenic mutations reported in VSX1 resulting in keratoconus.
The purpose of this study to perform mutational screening of the VSX1 gene in patients with sporadic and familial keratoconus in a European population and, for the first time, report the mutational analysis of the two newly identified VSX1 exons.
Methods
Patients and clinical assessment
Patients of European decent were recruited from the Royal Victoria Hospital, Belfast, and from the Department of Ophthalmology, University of Toronto. We certify that all applicable institutional and governmental regulations with regard to the ethical use of human volunteers were followed during this research. A diagnosis of definitive keratoconus was based on slit-lamp biomicroscopy, retinoscopy, and corneal topography. A history of penetrating keratoplasty or corneal surgery for keratoconus was considered as evidence of a definitive diagnosis, and pre-surgical clinical data were retrospectively assessed to ensure diagnostic accuracy. Corneal topography (videokeratography) was performed using the Topographic Modelling System (TMS-2) (Computed Anatomy Inc., New York, NY, USA) according to standardised methods previously reported by our group34 and was analysed for features diagnostic of keratoconus. Subjects with keratoconus were classified as familial cases if at least two or more individuals in one family met the clinical definition of keratoconus. When determining the familial segregation of VSX1 sequence variants, all available family members underwent a clinical and topographic examination to determine their clinical status before molecular analysis.
DNA extraction and sequencing
Genomic DNA was isolated from peripheral blood leukocytes using a Puregene blood extraction kit (Gentra Systems, Minneapolis, MN, USA) according to the manufacturer's protocol. Individual DNA samples were quantified using Nanodrop-1000 (Nanodrop Technologies, Wilmington, DE, USA) and a PCR dilution of 50 ng/μl was carried out for each DNA sample.
The coding exons, exon–intron junctions, and the 5′ and 3′ untranslated regions (UTRs) of VSX1 were amplified with at least 50 bp flanking the coding region of each exon. The five previously known exons, including the 3′ end of exon 3 specific to transcript variant 2 (exon 3b), were identical to those previously used,35 with modifications in PCR conditions. The primer pairs for the two novel exons,33 exons 6 and 7, were designed using Primer Detective V1.01 (Clontech Labs Inc., Cambridge, UK): exon 6 (5′-GCTTTTTCCTTAGGTTTAATCG-3′ and 5′-TGCTGCATGGGTCCATTTGTC-3′) with a PCR product of 310 bp and exon 7 (5′-TTCACTAAGCCACAGTCTCTCG-3′ and 5′-ACTTCAGCCCAGGAGTTAGAGG-3′) with a PCR product of 738 bp. The following additional sequencing primers were used for exon 7: 5′-GAGTTGAAAGCATTGGTCTCCC-3′ and 5′-AGGATGACACAGGAAAATCGGC-3′. NCBI dbSNP was screened to ensure no known SNPs were located in the primer-annealing site.
PCR amplification was carried out in a final reaction volume of 10 μl with 50 ng of genomic DNA, 10 × PCR buffer with 15 mm mgcl2, 5 × Q-solution (Qiagen, Sussex, UK), 200 μm of each dNTPs, 0.3 μm of gene-specific forward and reverse primers, and 2.5 units of HotStar Taq DNA polymerase (Qiagen). Thermal cycling was carried out in a 2700 ABI thermal cycler (Applied Biosystems, Foster City, CA, USA) under the following conditions: initial denaturation at 95°C for 15 min, followed by 40 cycles of 94°C for 30 s; 58°C (exon 1), 59°C (exons 2, 4, and 5), 62°C (exon 3), 61°C (exons 6 and 7) for 30 s, and 72°C for 30 s; and a final extension of 72°C for 10 min. The amplified PCR products were directly sequenced in a final reaction volume of 10 μl using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) and BetterBase diluent (Microzone Ltd, Sussex, UK). The cycling conditions were 94°C for 2 min, followed by 25 cycles of 94°C for 30 s, 55°C for 30 s, and 60°C for 4 min, with a final holding temperature at 4°C. Amplified DNA fragments were sequenced using both forward and reverse primers and were analysed on an ABI PRISM 3100 DNA sequencer (Applied Biosystems). Sequencing results were analysed manually and using the sequence analysis software Sequencher 4.7 (Gene Code Corporations, Suite, Ann Arbor, MI, USA).
Identified sequence variants were described according to the guidelines published by the Human Genome Variation Society. A total of 100 unrelated individuals (200 chromosomes) without ocular disease from a Northern Irish population were used as normal controls and VSX1 was sequenced in both directions in all individuals.
Bioinformatics
To determine the pathogenicity of the identified amino-acid substitutions, conservation was analysed using multiple sequence alignment with ClustalX and was visualised using GeneDoc software. Deleterious structural effects of amino-acid substitutions on protein function were assessed using the PolyPhen (Polymorphism Phenotyping) programme.36 Intronic changes and sequence variants close to splice sites not seen in controls were assessed to determine their impact of splicing signals using four scoring algorithms: Consensus Sequence, Neural Network, Information Theory, and Maximum Entropy.37, 38, 39, 40 To determine the possible mutational effects of synonymous sequence variants not seen in controls on exonic splicing, enhancers and silencers were modelled using servers ESEfinder41 and ESRsearch.42, 43
RNA extraction and RT-PCR
Total RNA was extracted from whole-blood samples from individuals harbouring VSX1 sequence variants with potential effects on splicing accuracy that were absent in control individuals. VSX1 gene expression can be detected in normal blood44 and hence RNA was extracted using a red blood cell lysis solution (Puregene, Gentra Systems) and an RNA extraction kit (TRIzol; Invitrogen, Paisley, UK), according to the manufacturer's instructions. Puromycin was used to inhibit nonsense-mediated mRNA decay45 and a DNase treatment was performed. To analyse the effect of novel silent mutation p.S263S on alternative splicing in a sporadic keratoconus patient, 1 μg of total RNA was reverse transcribed using random hexamer primers and reverse transcriptase (Sensiscript, Qiagen). RT-PCR was carried out using primers 5′-ATGCTGGCTGTGAAAACTGAGC-3′ and 5′-CTTCCTGGCTTCCTTA-TCATCC-3′ in exons 3 and 5, flanking the region of interest of VSX1 mRNA harbouring the synonymous p.S263S variant. RT-PCR products from the sporadic keratoconus patient harbouring p.S263S and control samples were assessed for aberrant splicing by gel electrophoresis.
Results
The entire coding region, exon–intron junctions, and 5′ and 3′UTR of VSX1 including two novel exons33 were analysed for mutations in a total of 66 unrelated patients with keratoconus from a European population (27 with familial keratoconus; 39 with sporadic keratoconus). Table 1 summarises all VSX1 sequence variants identified in this European cohort with keratoconus. Four sequence variants were not observed in 100 healthy controls: c.432C>G (p.D144E), c.479G>A (p.G160D), c.789C>T (p.S263S), and an intronic change c.844-13T>A (numbered with respect to NM_014588).
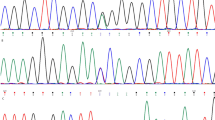
The variant p.D144E was detected in a family with keratoconus, although segregation was not observed (Figure 1). This sequence variant (p.D144E) was not observed in two siblings affected with keratoconus on the basis of clinical examination and topography, but was observed in a normal sibling and parent, although it was absent in 100 control individuals. Two sporadic keratoconus patients had the non-synonymous p.G160D change, which was not seen in controls (Figure 2a). The G160 residue is not well conserved across species (Figure 2b) and is predicted to be a benign change using the PolyPhen algorithm.

Sequence chromatogram of VSX1 c.432C>G (p.D144E) and segregation analysis. (a) DNA sequence chromatogram of the c.432C>G (p.D144E) sequence variant in VSX1 exon 2 (NM_014588), resulting in the substitution of aspartic acid (GAC: Asp; D) with glutamic acid (GAG: Glu; E), both acidic-charged polar amino-acid residues. (b) Segregation of p.D144E in familial keratoconus. Blackened symbols (II:1 and II:3) refer to affected individuals, whereas unblackened symbols are unaffected relatives (I:1, I:2, II:2, and II:4) (drawn using Cyrillic 2.1). Two unaffected individuals are heterozygous for p.D144E (I:1 and II:2), whereas affected family members with keratoconus are wild type, showing that p.D144E does not segregate with disease status.

Sequence chromatogram of VSX1 c.479G>A (p.G160D) and conservation analysis. (a) DNA sequence chromatogram of the c.479G>A (p.G160D) sequence variant in VSX1 exon 2 (NM_014588), resulting in the substitution of glycine (GGC: Gly; G) with aspartic acid (GAC: Asp; D), replacing a non-conservative substitution of a non-polar amino acid (glycine) with a charged polar residue (aspartic acid). (b) Multiple sequence alignment using ClustalX visualised using GeneDoc software for the glycine residue substituted in p.G160D showing 60% conservation across VSX1 from different species.
One sporadic keratoconus patient showed a novel silent mutation (p.S263S) in exon 4, which was not seen in 200 control chromosomes. The absence of this synonymous sequence variant in controls prompted a further analysis to assess its potential pathogenicity. Specifically, the ability of p.S263S to initiate mis-splicing events by altering putative ESE-binding motifs in the VSX1 coding sequence was modelled using ESEfinder41 and ESRsearch.42, 43 The introduction of p.S263S (c.789C>T; NM_014588) completely abolishes a potential motif for the SR protein, SF2/ASF, and causes a reduction in the binding motif for the SR protein, SC35. RT-PCR failed to show aberrant splicing around exon 4 in the presence of p.S263S (data not shown). One patient with familial keratoconus showed a sequence variant, c.844-13T>A, in intron 4 within the 3′ splice junction, which was not observed in 200 control chromosomes. This sequence change was predicted using bioinformatics to have an impact on splice site strength; however, segregation was not seen in the family (data not shown).
The known SNPs, rs8123716 (c.18 G>T; p.S6S), rs12480307 (c.546A>G; p.A182A), rs6138482 (c.650G>A; p.R217H), and rs743018 (c.843+140 C>T for DQ854809 or c.662+140 C>T for DQ854810 transcripts), were also found in keratoconus patients and failed to show segregation in familial cases. Two sequence variants in the 3′UTR were seen in sporadic and familial keratoconus patients. The sequence variant c.*28G>T segregated in two keratoconus families but was also seen in two healthy controls. The c.*50G>A change segregated in two keratoconus families but not in a third, and was also seen in six controls. The following polymorphic variants were seen in keratoconus patients and controls: c.711T>A (NM_199425; p.P237P), c.844-5-6insT (NM_014588), c.*28G>T (DQ854811/DQ854812), and c.*50G>A (DQ854809/DQ854810). No other previously reported pathogenic or potentially sequence variants were detected in the study cohort.
Discussion
To date, VSX1 remains the only major genetic factor to be identified in keratoconus pathogenesis. A number of protein coding changes were originally reported as being potentially pathogenic (p.L17P, p.D144E, p.L159M, p.G160D, p.R166W, p.H244R, and p.P247R),23, 35, 46 and a functional impact of p.R166W on homeodomain binding has been shown.23 However, there has been debate in literature with regard to the significance of VSX1 in keratoconus development.23, 29, 30, 35, 47 The frequency of VSX1 mutations in keratoconus study cohorts is variable, with some authors questioning the role of VSX1 in keratoconus.29, 30
Furthermore, the VSX1 knockout mouse did not have a corneal phenotype on light microscopy48 and the initial report of VSX1 expression in normal adult human cornea32 has not been replicated in subsequent studies on normal and keratoconic corneas.23, 49, 50 VSX1 expression was detected at a low level in murine cornea51, 52, 53 and was recently reported in human neonatal cornea.33 There is experimental evidence that VSX1 has a role in corneal wound healing, in which it participates in the differentiation of corneal keratocytes into myofibrobalsts,54 explaining a potential biological role for its involvement in keratoconus pathogenesis.
VSX1 mutations have been reported in 4.7% (3 of 63)23 and 8.75% (7 of 80) of unrelated keratoconus patients,35 whereas other groups have failed to identify VSX1 pathogenic sequence variants in keratoconus.29, 30, 47 Mutations in VSX1 have also been reported in PPCD1 (MIM#122000)23, 46 and in combination with keratoconus.23 A p.A256S VSX1 mutation was reported in an African-American family with corneal endothelial abnormalities, craniofacial anomalies, and abnormalities in retinal and auditory bipolar cells.55
Recently, two novel exons downstream of the original VSX1 gene sequence were identified and VSX1 is now known to consist of seven exons with a complex splicing pattern producing a total of six transcripts.33 The aim of this study was to screen the complete gene sequence of VSX1 and to determine the contribution of the novel VSX1 sequence to keratoconus pathogenesis.
The p.G160D variant was detected in two sporadic keratoconuses and was absent in 100 normal control individuals. This change was reported as being potentially pathogenic in posterior polymorphous dystrophy23 on the basis of segregation and absence from 277 control individuals. Héon et al23 reported that the G160 residue is not highly conserved in keeping with the multiple sequence alignment illustrated in Figure 2b. Bisceglia et al35 detected p.G160D in two families with keratoconus. In one family, p.G160D was detected in a patient with keratoconus, in two relatives with topographically suspected keratoconus, but was not seen in two further relatives with topographically suspected keratoconus. In the second family, p.G160D was present in a compound heterozygote with p.L17P and resulted in clinical keratoconus. Two other family members had p.L17P alone and had evidence of topographically suspected keratoconus. The change p.L17P was also reported as pathogenic in a sporadic case of keratoconus and was absent from 125 control individuals. Bisceglia et al35 evaluated the segregation of p.G160D in family members with subclinical or ‘forme fustre’ keratoconus and it is difficult therefore to determine its true role in keratoconus pathogenesis. The pathogenicity of p.G160D can be debated from this evidence and it may represent a rare polymorphism or genetic modifier. Compound heterozygotes with p.G160D in association with p.L17P35 and p.P247R23 had clinically more severe corneal phenotypes.
The p.D144E variant was previously reported as disease causing in two affected family members with PPCD1 and keratoconus,23 but has been detected in healthy controls56 and glaucoma probands,23 as well as in keratoconus patients.29, 35, 47 In the cases with p.D144E reported by Bisceglia et al35 from keratoconus families, one case was effectively a sporadic patient and in the second family in which segregation was reported, three out of the four affected patients had a diagnosis of forme fustre or subclinical keratoconus. Recently, p.D144E was shown not to segregate with the disease phenotype in a family with clinical keratoconus.47 Similarly, in this study, p.D144E was detected in a family with keratoconus, but segregation was not detected. Cumulative evidence suggests that p.D144E is a non-pathogenic polymorphism. Determining the pathogenicity of reported sequence variants has been assisted by screening familial keratoconus cohorts and assessing segregation.35, 47
Two other sequence variants were detected in this study but were absent in controls. The intronic change c.844-13T>A, although predicted to have a significant impact on splice site strength, did not segregate in a family with keratoconus. The synonymous variant p.S263S was seen in a sporadic patient with keratoconus, although it was absent in controls. Synonymous or translationally silent sequence changes have the potential to alter the efficiency and specificity of alternative splicing.57 The recent elucidation of the diverse transcript profile of VSX133 and the predicted effect of p.S263S on putative ESEs led us to evaluate the ability of this sequence variant to induce aberrant splicing. RNA analysis failed to detect aberrant VSX1 transcript processing in the presence of p.S263S.
In summary, a comprehensive screening of the VSX1 gene, including two newly identified exons in patients with sporadic and familial keratoconus, in a European population confirms that VSX1 has a minor role in keratoconus pathogenesis and further study is required to determine the molecular genetic basis of this significant corneal disorder.
References
Cosar CB, Sridhar MS, Cohen EJ, Held EL, Alvim Pde T, Rapuano CJ et al. Indications for penetrating keratoplasty and associated procedures, 1996–2000. Cornea 2002; 21: 148–151.
Kang PC, Klintworth GK, Kim T, Carlson AN, Adelman R, Stinnett S et al. Trends in the indications for penetrating keratoplasty, 1980–2001. Cornea 2005; 24: 801–803.
Rabinowitz YS . Major review: keratoconus. Surv Ophthalmol 1998; 42: 297–319.
Falls HF, Allen AW . Dominantly inherited keratconus: report of a family. J Genet Hum 1969; 17: 317–324.
Parker J, Ko WW, Pavlopoulos G, Wolfe PJ, Rabinowitz YS . Videokeratopography of keratoconus in monozygotic twins. J Refract Surg 1996; 12: 180–183.
Wang Y, Rabinowitz YS, Rotter JI, Yang H . Genetic epidemiological study of keratoconus: evidence for major gene determination. Am J Med Genet 2000; 93: 403–409.
Rabinowitz YS, Maumenne IH, Lundergan MK, Puffenberger E, Zhu D, Antonarakis S et al. Molecular genetic analysis in autosomal dominant keratoconus. Cornea 1992; 11: 302–308.
Edwards M, McGhee CN, Dean S . The genetics of keratoconus. Clin Experiment Ophthalmol 2001; 29: 345–351.
Tyynismaa H, Sistonen P, Tuupanen S, Tervo T, Dammert A, Latvala T et al. A locus for autosomal dominant keratoconus: linkage to 16q22.3-q23. Invest Ophthalmol Vis Sci 2002; 43: 3160–3164.
Rabinowitz YS, Rasheed K . KISA% index: a quantitative videokeratography algorithm embodying minimal topographic criteria for diagnosing keratoconus. J Cataract Refract Surg 1999; 25: 1327–1335.
Rao SN, Raviv T, Majmudar PA, Epstein RJ . Role of Orbscan II in screening keratoconus suspects before refractive corneal surgery. Ophthalmology 2002; 109: 1642–1646.
Klyce SD, Smolek MK, Maeda N . Keratoconus detection with the KISA% method-another view. J Cataract Refract Surg 2000; 26: 472–474.
Maeda N, Klyce SD, Smolek MK . Comparison of methods for detecting keratoconus using videokeratography. Arch Ophthalmol 1995; 113: 870–874.
Claude S, Verdier R, Arnaud B, Schmitt-Bernard CF . Accuracy of videokeratographic quantitative criteria for detection of keratoconus suspects in families with keratoconus. J Fr Ophtalmol 2004; 27: 773–778.
Levy D, Hutchings H, Rouland JF, Guell J, Burillon C, Arné JL et al. Videokeratographic anomalies in familial keratoconus. Ophthalmology 2004; 111: 867–874.
Hutchings H, Ginisty H, Le Gello M, Levy D, Stoësser F, Rouland JF et al. Identification of a new locus for isolated familial keratoconus at 2p24. J Med Genet 2005; 42: 88–94.
Brancati F, Valente EM, Sarkozy A, Feher J, Del Duca D, Mingarelli R et al. A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J Med Genet 2004; 41: 188–192.
Rabinowitz YS, Rasheed K, Yang H, Elashoff J . Accuracy of ultrasonic pachymetry and videokeratography in detecting keratocnonus. J Cataract Refract Surg 1998; 24: 196–201.
Rabinowitz YS . Keratoconus. Surv Ophthalmol 1998; 42: 297–319.
Rabinowitz YS . The genetics of keratoconus. Ophthalmol Clin North Am 2003; 16: 607–620, vii.
Vincent AL, Rootman D, Munier FL, Héon E . A molecular perspective on corneal dystrophies. Dev Ophthalmol 2003; 37: 50–66.
Heon E, Mathers WD, Alward WL, Weisenthal RW, Sunden SL, Fishbaugh JA et al. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet 1995; 4: 485–488.
Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G et al. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 2002; 11: 1029–1036.
Hughes AE, Dash DP, Jackson AJ, Frazer DG, Silvestri G . Familial keratoconus with cataract: linkage to the long arm of chromosome 15 and exclusion of candidate genes. Invest Ophthalmol Vis Sci 2003; 44: 5063–5066.
Dash DP, Silvestri G, Hughes AE . Fine mapping of the keratoconus with cataract locus on chromosome 15q and candidate gene analysis. Mol Vis 2006; 12: 499–505.
Tang YG, Rabinowitz YS, Taylor KD, Li X, Hu M, Picornell Y et al. Genomewide linkage scan in a multigeneration Caucasian pedigree identifies a novel locus for keratoconus on chromosome 5q14.3-q21.1. Genet Med 2005; 7: 397–405.
Fullerton J, Paprocki P, Foote S, Mackey DA, Williamson R, Forrest S . Identity-by-descent approach to gene localisation in eight individuals affected by keratoconus from north-west Tasmania, Australia. Hum Genet 2002; 110: 462–470.
Li X, Rabinowitz YS, Tang YG, Picornell Y, Taylor KD, Hu M et al. Two-stage genome-wide linkage scan in keratoconus sib pair families. Invest Ophthalmol Vis Sci 2006; 47: 3791–3795.
Aldave AJ, Yellore VS, Salem AK, Yoo GL, Rayner SA, Yang H et al. No VSX1 gene mutations associated with keratoconus. Invest Ophthalmol Vis Sci 2006; 47: 2820–2822.
Tang YG, Picornell Y, Su X, Li X, Yang H, Rabinowitz YS . Three VSX1 gene mutations, L159M, R166W, and H244R, are not associated with keratoconus. Cornea 2008; 27: 189–192.
Hayashi T, Huang J, Deeb SS . RINX(VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics 2000; 67: 128–139.
Semina EV, Mintz-Hittner HA, Murray JC . Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics 2000; 63: 289–293.
Hosseini SM, Herd S, Vincent AL, Héon E . Genetic analysis of chromosome 20-related posterior polymorphous corneal dystrophy: genetic heterogeneity and exclusion of three candidate genes. Mol Vis 2008; 14: 71–80.
Burns MD, Johnston FM, Frazer DG, Patterson C, Jackson AJ . Keratoconus: an analysis of corneal asymmetry. Br J Ophthalmol 2004; 88: 1252–1255.
Bisceglia L, Ciaschetti M, De BP, Campo PA, Pizzicoli C, Scala C et al. VSX1 mutational analysis in a series of Italian patients affected by keratoconus: detection of a novel mutation. Invest Ophthalmol Vis Sci 2005; 46: 39–45.
Sunyaev S, Ramensky V, Koch I, Lathe III W, Kondrashov AS, Bork P . Prediction of deleterious human alleles. Hum Mol Genet 2001; 10: 591–597.
Shapiro MB, Senapathy P . RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987; 15: 7155–7174.
Brunak S, Engelbrecht J, Knudsen S . Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 1991; 220: 49–65.
Yeo G, Burge CB . Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11: 377–394.
Nalla VK, Rogan PK . Automated splicing mutation analysis by information theory. Hum Mutat 2005; 25: 334–342.
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR . ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003; 31: 3568–3571.
Fairbrother WG, Yeh RF, Sharp PA, Burge CB . Predictive identification of exonic splicing enhancers in human genes. Science 2002; 297: 1007–1013.
Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I et al. Comparative analysis identifies exonic splicing regulatory sequences—The complex definition of enhancers and silencers. Mol Cell 2006; 22: 769–781.
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 2004; 101: 6062–6067.
Noensie EN, Dietz HC . A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat Biotechnol 2001; 19: 434–439.
Valleix S, Nedelec B, Rigaudiere F, Dighiero P, Pouliquen Y, Renard G et al. H244R VSX1 is associated with selective cone ON bipolar cell dysfunction and macular degeneration in a PPCD family. Invest Ophthalmol Vis Sci 2006; 47: 48–54.
Liskova P, Ebenezer ND, Hysi PG, Gwilliam R, El-Ashry MF, Moodaley LC et al. Molecular analysis of the VSX1 gene in familial keratoconus. Mol Vis 2007; 13: 1887–1891.
Chow RL, Volgyi B, Szilard RK, Ng D, McKerlie C, Bloomfield SA et al. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc Natl Acad Sci USA 2004; 101: 1754–1759.
Nielsen K, Birkenkamp-Demtroder K, Ehlers N, Orntoft TF . Identification of differentially expressed genes in keratoconus epithelium analyzed on microarrays. Invest Ophthalmol Vis Sci 2003; 44: 2466–2476.
Rabinowitz YS, Dong L, Wistow G . Gene expression profile studies of human keratoconus cornea for NEIBank: a novel cornea-expressed gene and the absence of transcripts for aquaporin 5. Invest Ophthalmol Vis Sci 2005; 46: 1239–1246.
Chow RL, Snow B, Novak J, Looser J, Freund C, Vidgen D et al. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech Dev 2001; 109: 315–322.
Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet 2005; 77: 694–708.
Hayashi T, Huang J, Deeb SS . Expression of rinx/vsx1 during postnatal eye development in cone-bipolar, differentiating ganglion, and lens fiber cells. Jpn J Ophthalmol 2005; 49: 93–105.
Barbaro V, Di IE, Ferrari S, Bisceglia L, Ruzza A, De Luca M et al. Expression of VSX1 in human corneal keratocytes during differentiation into myofibroblasts in response to wound healing. Invest Ophthalmol Vis Sci 2006; 47: 5243–5250.
Mintz-Hittner HA, Semina EV, Frishman LJ, Prager TC, Murray JC . VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology 2004; 111: 828–836.
Aldave AJ, Yellore VS, Principe AH, Abedi G, Merrill K, Chalukya M et al. Candidate gene screening for posterior polymorphous dystrophy. Cornea 2005; 24: 151–155.
Cartegni L, Chew SL, Krainer AR . Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3: 285–298.
Acknowledgements
We extend our gratitude to the patients who participated in this study. This study was funded by the Research and Development Office, Northern Ireland RRG Grant: 4.46.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the Annual Congress of the Royal College of Ophthalmologists 2009, and in part at The Association for Research in Vision and Ophthalmology (ARVO) 2008
Web Resources
NCBI dbSNP:
http://www.ncbi.nlm.nih.gov/projects/SNP/
Human Genome Variation Society:
ClustalX:
ftp://ftp.ebi.ac.uk/pub/software/clustalw2
GeneDoc:
http://www.nrbsc.org/gfx/genedoc/index.html
PolyPhen (Polymorphism Phenotyping):
http://genetics.bwh.harvard.edu/pph/
Consensus Sequence:
http://www.genet.sickkids.on.ca/~ali/splicesitescore.html
Neural Network:
http://www.fruitfly.org/seq_tools/splice.html
Information Theory:
Maximum Entropy: http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
ESE finder:
http://rulai.cshl.edu/tools/ESE/
ESRsearch:
Rights and permissions
About this article
Cite this article
Dash, D., George, S., O'Prey, D. et al. Mutational screening of VSX1 in keratoconus patients from the European population. Eye 24, 1085–1092 (2010). https://doi.org/10.1038/eye.2009.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2009.217
Keywords
This article is cited by
-
Whole exome sequencing highlights variants in association with Keratoconus in Jordanian families
BMC Medical Genetics (2020)
-
The Proteins of Keratoconus: a Literature Review Exploring Their Contribution to the Pathophysiology of the Disease
Advances in Therapy (2019)
-
Genetic Aspects of Keratoconus: A Literature Review Exploring Potential Genetic Contributions and Possible Genetic Relationships with Comorbidities
Ophthalmology and Therapy (2018)
-
Identification of novel pathogenic variants and novel gene-phenotype correlations in Mexican subjects with microphthalmia and/or anophthalmia by next-generation sequencing
Journal of Human Genetics (2018)
-
Analysis of the VSX1 gene in sporadic keratoconus patients from China
BMC Ophthalmology (2017)
