
Abstract
Atopic dermatitis (AD) is a chronic inflammatory skin disease, and its prevalence is increasing. AD usually elicits skin barrier dysfunction, dry skin and itching. As the mechanisms of AD remain unknown, there is an urgent need to find effective therapies. Because of the diversity and complexity of marine environments, the discovery of drugs from marine organisms as novel therapeutic agents for human diseases has seen renewed interest. Dihydroaustrasulfone alcohol (WA-25), the synthetic precursor of austrasulfone, which is a natural product isolated from a Formosan soft coral, has been shown to possess many therapeutic effects in our previous studies. However, the detailed mechanisms and therapeutic effects of WA-25 on AD are incompletely understood. We performed in vitro and in vivo studies to examine the effects of WA-25 on AD. We showed that WA-25 blocks inflammation and oxidative stress. Simultaneously, we also found that WA-25 reduces the AD scores and AD-induced transepidermal water loss (TEWL), scratching behavior, and alloknesis. WA-25 is more effective in cases of AD than are the drugs that are currently used clinically. Importantly, we also found that when nucleophosmin (NPM) was inhibited or when its expression was reduced, the anti-inflammatory and anti-AD effects of WA-25 were blocked. These data suggest that NPM plays dual roles in inflammation and AD. Overall, these results suggest that WA-25 is a potential anti-inflammatory and AD therapeutic agent that is modulated by NPM.
Similar content being viewed by others


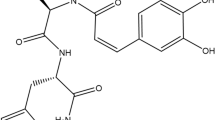
Introduction
Atopic dermatitis (AD), also known as atopic eczema, is a chronic inflammatory skin disease whose prevalence is increasing globally.1 AD is often accompanied by skin thickening; infiltration of inflammatory cells, such as macrophages, lymphocytes and mast cells; skin barrier dysfunction; and increased transepidermal water loss (TEWL) in areas with lesions.2 Moderate lesions require moisturizing products and either calcineurin inhibitors or topical steroid therapy.3 Many corticosteroids have been used to treat AD over the years.4 Among them, hydrocortisone was used first, and its efficacy in treating AD has been known for 60 years.5 Hydrocortisone creams are also still used as a reference drug for AD.6, 7 However, the currently available therapies have limited success, do not typically affect the rate of recurrence, and have considerable side effects. The detailed mechanism of AD is not entirely clear, but induction may involve certain proinflammatory factors such as tumor necrosis factor-alpha (TNF-α), inducible nitric oxide synthase (iNOS) and chemokines.8
Disorders of the skin barrier are also found in early AD. Many studies have indicated that an abnormal skin barrier is associated with pro-filaggrin (FLG) in AD lesions.9 These findings suggest a possible explanation for why patients who suffer from AD have increased TEWL. 2,4-dinitrochlorobenzene (DNCBs) are often used to induce AD in rodents and have been found to upregulate inflammatory proteins in macrophages. Therefore, a DNCB-induced AD model has been developed to assess the efficacy of possible treatments.10 For example, the fish oil derivative resolvin E1 was also shown to block inflammation in macrophages and to alleviate AD in mice.11 Based on the above findings, by screening substances for their anti-inflammatory activity in macrophages, new treatments for AD can be identified and developed.
Macrophages play an important role in innate immunity. These phagocytic cells maintain homeostasis and help clear debris and toxic substances.12 Macrophages can be activated by lipopolysaccharide (LPS), interferon-γ, and cytokines. LPS can activate mouse macrophages (RAW 264.7 cells) via the Toll-like receptor 4, thereby causing inflammation. Active macrophages release certain inflammatory factors such as TNF-α, interleukin 6 and iNOS.13 Heme oxygenase-1 (HO-1) is the rate-limiting enzyme for the catalysis of heme into biliverdin, carbon monoxide and iron.14 Over the past decade, HO-1 has been shown to block inflammation, oxidation and apoptosis.15 Furthermore, the induction of HO-1 expression in macrophages is essential for its anti-inflammatory effects.16 Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) also plays a vital role in the regulation of immunity and inflammation.17 Some reports have indicated that NF-κB interacts with NPM, which is considered an alarmin that shuttles between the nucleus and the cytoplasm.18 However, the exact relationship between NF-κB and NPM remains unknown.
Some scientists have acknowledged that drug discovery from marine organisms has a relatively high potential for success.19 Our previous studies indicated that extracts from the Formosan soft coral Cladiella australis have several bioactivities. WA-25, the synthetic precursor of austrasulfone, a natural product produced by C. australis, possesses anti-inflammatory activity and elicits therapeutic effects against neuropathic pain, atherosclerosis, multiple sclerosis and cancer.20, 21 Although the beneficial effects of WA-25 have been described for many diseases, the mechanism by which it modulates the progression of AD remains poorly understood. Therefore, we investigated the detailed anti-inflammatory and other therapeutic effects of WA-25 on AD.
Materials and methods
Chemicals
Dihydroaustrasulfone alcohol (WA-25) was originally designed and synthesized by Sheu and colleagues.21 WA-25 in this study was partially provided by the Research Center of National Research Program for Biopharmaceuticals, Taiwan (http://nrpb.sinica.edu.tw/zh-hant/rc). Penicillin, streptomycin, dimethyl sulfoxide, LPS (Escherichia coli), paraformaldehyde, DNCB, hydroxypropyl cellulose (HPC), Griess reagent, NPM, HO-1, sirtuin-6 (Sirt-6) and anti-β-actin antibody were obtained from Sigma Chemical (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), sodium pyruvate, L-glutamine, trypsin-EDTA, alamarBlue, cellular reactive oxygen species (ROS) detection assay kit, DAPI solution, Lipofectamine 3000 and green or red fluorescence-conjugated secondary antibodies were purchased from Invitrogen Co. (Grand Island, NY, USA). Anti-iNOS and anti-COX-2 antibodies and NSC 348884 were purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Anti-Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), anti-histone, anti-E-cadherin, anti-FLG-1, anti-histamine, anti-substance P, and anti-glial fibrillary acidic protein (GFAP) antibodies were obtained Abcam Corp. (Cambridge, UK). Anti-p65 NF-κB and anti-lysosomal-associated membrane protein 1 (LAMP-1) antibodies were purchased from Millipore Corp. (Billerica, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Cell line culture
The growth conditions used for culturing the RAW 264.7 cell line were described previously.22 Briefly, RAW 264.7 macrophages (No. TIB-71) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM containing 10% heat-inactivated FBS, 2 mM glutamine, 1 mM pyruvate, 4.5 g l−1 glucose, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin. The cell cultures were maintained at 37 °C in a humidified atmosphere containing 5 CO2 and 95% air. Cells were trypsinized for subculturing in different-sized wells or dishes. Culture plastic ware was obtained from Corning (Corning, NY, USA). All experiments were performed overnight after cell seeding.
Anti-inflammatory assay
RAW 264.7 cells were seeded in 10-cm dishes at a density of 1 × 106 cells. Inflammation was induced by incubation in media containing only LPS (0.01 μg ml−1) without WA-25. For the anti-inflammatory activity assay, WA-25 was added to the cells. The cells were then washed with ice-cold phosphate-buffered saline (PBS), lysed in ice-cold lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 100 μg ml−1 phenylmethylsulfonyl fluoride and 1 μg ml−1 aprotinin), and centrifuged at 20 000 g for 30 min at 4 °C. The supernatants were decanted and reserved for western blotting. Protein concentrations were measured using the DC protein assay kit (Bio-Rad, Hercules, CA, USA).
Western blot analysis
Western blotting was performed according to the method described in our previous study.23 An equal volume of sample buffer (2% 2-mercaptoethanol, 2% sodium dodecyl sulfate (SDS), 0.1% bromophenol blue, 10% glycerol, and 50 mM Tris-HCl (pH 7.2)) was added to the samples, and the protein lysates were loaded onto a 10% SDS-polyacrylamide gel. Electrophoresis was carried out at 150 V for 90 min. After electrophoresis, gels were transferred overnight at 4 °C in transfer buffer (380 mM glycine, 50 mM Tris-HCl, 1% SDS and 20% methanol) onto a polyvinylidene difluoride membrane (PVDF; Immobilon-P, Millipore Corp. (0.45-μm pore size)). The PVDF membrane was first blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween (TTBS; 20 mM Tris-HCl, 0.1% Tween 20, and 137 mM NaCl (pH 7.4)) and incubated overnight at 4 °C with the primary antibodies. A horseradish peroxidase-conjugated secondary antibody was used for detection. The bound antibodies were detected by chemiluminescence (Millipore Corp.). The images were obtained using the UVP BioChemi Imaging System, and the LabWorks 4.0 software (UVP, Upland, CA, USA) was used to quantify the relative densities.
Immunocytochemistry
To examine the signaling proteins, RAW 264.7 cells were seeded onto slides at a density of 0.5–1 × 105 cells. Immunocytochemistry was performed using modified methods as previously described.24 After treatment, RAW 264.7 cells were fixed with 4% paraformaldehyde in PBS buffer for 8 min and washed three times with PBS buffer. The cells were blocked with 4% normal goat serum dissolved in PBS containing 0.01% Triton X-100. After blocking, the RAW 264.7 cells were washed three times with PBS buffer and then incubated overnight at 4 °C with primary antibodies. Cells were washed three times with PBS buffer, blocked as described previously, and then incubated for 2 h at 37 °C with green or red fluorescence-conjugated secondary antibody. Fluorescence was visualized using a Leica DM6000 CFS confocal microscope and analyzed with Leica Microsystems software (Leitz-Park, Wetzlar, Germany).
NO production and determination of intracellular ROS
Cells were seeded at a density of 105 cells per well in a 96-well plate. NO production was measured according to the Griess reaction. Intracellular ROS was examined using CellROX Green Reagent fluorescent dye (Invitrogen Co., Grand Island, NY, USA). The measurements were carried out as described previously.16 The absorbance was detected by an Epoch 2 Touch Screen (Bio-Tek, Winooski, VT, USA). The fluorescence was visualized using a Leica DM6000 microscope.
Induction of AD
This method was modified from a method used in previous studies.25 Male BALB/c mice (aged 6 weeks) were obtained from BioLASCO Co. Ltd., Taipei, Taiwan, and used for the induction of AD. The experimental procedure is shown schematically in Figure 2a. Briefly, DNCB was diluted in a mixture of acetone and olive oil (4:1). The DNCB solution (50 μl of 1% DNCB) was applied to the shaved backs of mice three times a week. WA-25 was prepared as a gel and used for treatment from days 7 to 15. All animals were sacrificed on day 15, and samples were collected for further analysis. The AD score was evaluated according to symptoms such as (a) scaling, (b) erythema, (c) erosion and (d) edema. Each symptom was scored as 0 (no symptom), 1 (mild), 2 (moderate), or 3 (severe). We followed the Guiding Principles in the Care and Use of Animals of the American Physiology Society, and all experiments were approved by the National Sun Yat-sen University Institutional Animal Care and Use Committee.
Preparation of WA-25 gel
A 0.1% (w/w) WA-25 gel was used in this study and was prepared with HPC, distilled water and ethanol, as described in our previous report.26 The solution was stirred overnight until a clear gel was formed. The gel was prepared fresh before the experiments.
Measurement of TEWL
TEWL was detected on the dorsal skin using a Tewameter (TM300; Courage and Khazawa, Cologne, Germany) following the method described in a previous report.27 TEWL readings were recorded after stabilization, and the data were expressed in g m−2 h−1. TEWL was measured on day 15 before the animals were killed.
Evaluation of scratching behavior
The scratching behavior test was performed as previously described.28 All mice were acclimated in acrylic cages for ~15 min. After acclimation, the mice were observed for scratching behavior for 30 min.
Alloknesis assay
Alloknesis was assessed as previously described.29 Briefly, a von Frey filament (0.7 mN) was applied to the skin for 30 min. If the mouse scratched the site where the von Frey filament was applied, it was defined as a positive response. The alloknesis score was calculated as the total number of scratches in 30 min.
Immunohistochemistry
Paraffin-embedded mouse skin sections were deparaffinized in xylene and rehydrated in a graded ethanol series. Slides were blocked with normal goat serum for 40 min and then incubated with primary antibody. After washing, slides were incubated first with biotinylated secondary antibody and then with the DAB substrate (Vector Laboratory, Burlingame, CA, USA) for 1–3 min, followed by counterstaining with hematoxylin for 5–10 s. All images were captured by a Leica DM 6000B light microscope with a SPOT Idea 5 MP Scientific Digital Camera System (Diagnostic Instruments Inc., Sterling Heights, MI, USA). Some skin and spinal cord tissues were collected and cut in a cryostat for immunohistochemistry. Briefly, the tissue sections were blocked with normal goat serum and incubated with primary antibodies overnight. The sections were then incubated for 1 h at room temperature with green or red fluorescence-conjugated secondary antibody. The images were visualized using a Leica DM6000 CFS confocal microscope and analyzed using the Leica Microsystems software.
siRNA in vitro and in vivo applications
SMARTpool: Accell Npm1 siRNA was synthesized by Dharmacon Research (Thermo Fisher Scientific, Lafayette, CO, USA). For knockdown in RAW 264.7 cells, Npm1 siRNA was added to the medium with Lipofectamine 3000 overnight, and an anti-inflammatory assay was then performed. The topical siRNA formulation was prepared as a gel by mixing SMARTpool: Accell Npm1 siRNA using a modified protocol from Hegde et al.30 We applied the siRNA mixture to the skin of the mice once a day for 14 consecutive days.
Inflammation antibody array
Inflammation antibody array membranes (Abcam, Cambridge, MA, USA) were used to examine the profiles of inflammatory cytokines in mouse serum following the manufacturer’s instructions. Spots were detected by chemiluminescence using the UVP BioChemi Imaging System and LabWorks 4.0 software (UVP, Upland, CA, USA).
Data and statistical analysis
All data were presented as the means±s.e.’s of the means. For immunoblot analyses, the intensity of the experimental results was determined as the integrated optical density and compared to the average optical density of the corresponding control group. For statistical analysis, all data were analyzed by variance, along with Dunnett’s test for multiple comparisons. Differences were significant when the P-value was <0.05.
Results
Effects of WA-25 on inflammation and oxidative stress in LPS-stimulated macrophages
We investigated whether WA-25 has anti-inflammatory effects, using LPS-stimulated macrophages. Treatment with LPS alone markedly increased the iNOS protein expression levels, which is a proinflammatory cytokine, whereas treatment with both LPS and WA-25 significantly inhibited the iNOS level (Figure 1a). We further found that WA-25 significantly decreased LPS-induced NO production (Figure 1b). To determine whether WA-25 blocks the effects of oxidative stress, we showed that WA-25 can inhibit the LPS-induced upregulation of ROS (Figure 1c). In agreement with these observations, we found no expression of HO-1 in the presence or absence of LPS, whereas WA-25 increased HO-1 expression (Figure 1d and e). Furthermore, the level of Nrf2, which is upstream of HO-1, decreased after LPS induction and was upregulated significantly after incubation with WA-25 (Figure 1e).

Anti-inflammatory and anti-oxidative stress effects of WA-25 in LPS-stimulated macrophages after 16 h. (a) Western blot of iNOS from LPS-stimulated macrophages treated with 10 μM WA-25. The relative density of iNOS is shown in the graphs. (b) NO production from LPS-stimulated macrophages treated with 10 μM WA-25. (c) Reactive oxygen species (ROS) production from macrophages in the control, LPS and LPS+WA-25 groups. (d) Immunostaining of HO-1 (red) in macrophages in the control, LPS and LPS+WA-25 groups. (e) Western blot analysis of HO-1 and Nrf2 from LPS-stimulated macrophages treated with 10 μM WA-25. Relative densities of HO-1 and Nrf2 are shown in the graphs under the blots. *Significantly (P<0.05) different from the LPS-stimulated group. Scale bar=30 μm; n=6 per group.
Therapeutic effects of WA-25 on AD
We induced AD in BALB/c mice with DNCB to evaluate whether WA-25 can alleviate AD phenotypes. Our experimental schedule for the induction of AD is shown in Figure 2a. We found that DNCB could induce AD lesions in mice on day 15 (Figure 2b). Treatment with WA-25 could relieve AD phenotypes better than treatment with hydrocortisone cream (HC) could. Additionally, WA-25 significantly improved the AD scores and inhibited AD-induced TEWL, scratching behavior and alloknesis (Figure 2c–f). Although treatment with HC could also significantly relieve these phenotypes and improve the AD scores, its effects were not as noticeable as those of WA-25. In addition, HC could not inhibit AD-induced alloknesis. Figure 2g shows that treatment with WA-25 reduced AD-induced iNOS overexpression.

Therapeutic effects of WA-25 on AD. (a) Protocol for induction of AD in BALB/c mice. (b) The photos of the control, AD, WA-25-treated (AD+W) and HC-treated (HC) groups were taken on day 15. (c) AD scores of the control, AD, AD+W and HC groups. (d) TEWL of the control, AD, AD+W and HC groups. (e) Scratching behavior of the control, AD, AD+W and HC groups. (f) Alloknesis scores of the control, AD, AD+W and HC groups. (g) Immunostaining of iNOS and HO-1 in the skin of animals from the control, AD and AD+W groups. *Significantly (P<0.05) different from the AD group. #Significantly (P<0.05) different from the AD+W group. Scale bar=50 μm. n=6 per group. AD, atopic dermatitis; TEWL, transepidermal water loss.
Interaction between NF-κB and NPM plays an important role in inflammation
To determine whether WA-25 can inhibit the iNOS-related pathway, we examined NF-κB activity, which is upstream of iNOS. As shown in Figure 3a, LPS induced NF-κB translocation from the cytoplasm into the nucleus. However, WA-25 significantly decreased the translocation of p65 NF-κB. Other studies have indicated that sirt-6 can inhibit NF-κB transcriptional activity.31 Importantly, we found that LPS could decrease sirt-6 protein expression in the nucleus, whereas WA-25 significantly restored sirt-6 expression (Figure 3b). To further examine how WA-25 regulates NF-κB signaling, we tried to clarify the pathways in which NPM might be involved. As shown in Figure 3c, LPS decreased the NPM levels in the nucleus, whereas treatment with WA-25 increased NPM expression. In addition, LPS slightly reduced the total NPM expression (Figure 3c). Interestingly, immunofluorescence staining of both p65 NF-κB and NPM showed that p65 NF-κB was almost exclusively expressed in the cytoplasm and that NPM was primarily expressed in the nucleus (Figure 3d). However, LPS stimulated the translocation of p65 NF-κB into the nucleus and of NPM into the cytoplasm. In addition, some NPM-NF-κB interactions were evident in the nucleus in both the control and WA-25 groups. To clarify why the total NPM levels decreased after LPS stimulation, we investigated whether NPM could be degraded by lysosomes. We used the lysosomal marker LAMP1 to study the association between NPM and lysosomes. As shown in Figure 3e, we found an accumulation of LAMP1 in the control and WA-25 groups. However, LPS induced NPM degradation, presumably by lysosomes, in the cytoplasm. Therefore, we proposed that WA-25 might regulate NPM translocation into the nucleus and binding to NF-κB to block inflammatory signaling. To confirm this hypothesis, we assessed whether WA-25 could regulate NPM binding to NF-κB. Using immunoprecipitation, we observed that WA-25 significantly increased NPM binding to NF-κB compared to the LPS group (Figure 3f).

Effects of WA-25 on NF-κB activity and NF-κB–NPM interaction after 6 h. (a) Western blot and ratio of p65 NF-κB translocation is shown. (b) Western blot analysis of sirt-6. Relative densities of sirt-6 protein expression are provided. (c) Western blot analysis of NPM. Relative densities of nuclear and total NPM protein expression are provided. (d) p65 NF-κB (green) and NPM (red) immunostaining. Scale bar=5 μm. (e) Immunostaining of LAMP-1 (green) and NPM (red). Scale bar=7.5 μm. (f) Immunoprecipitation of NPM and immunoblot analysis of p65 NF-κB. Relative densities of NPM and p65 NF-κB are provided. *Significantly (P<0.05) different from the LPS-stimulated group; n=6 per group.
Effects of WA-25 on inflammation regulated by NPM
To demonstrate that the effects of WA-25 on inflammation are regulated by NPM, we used both NSC 348884, an NPM protein inhibitor, and NPM siRNA to clarify the mechanisms involved. As shown in Figure 4a, the anti-inflammatory effects of WA-25 were inhibited by NSC 348884. Similarly, NPM knockdown suppressed these anti-inflammatory effects (Figure 4b). We then asked whether the therapeutic effects of WA-25 on AD could also be achieved by NPM. As shown in Figure 4c, the therapeutic effects of WA-25 on AD were inhibited by both NSC 348884 and NPM siRNA. Concurrently, the AD score, AD-induced TEWL, scratching behavior, and alloknesis, which were all improved by the WA-25 treatment, were disrupted by inhibiting the NPM protein (Figure 4d–g). To clarify the detailed mechanisms involved, we chose the NPM siRNA group for the following experiments. As shown in Figure 4h, NPM was primarily located in the nucleus in the control group but was translocated into the cytoplasm in the AD group. However, treatment with WA-25 induced the translocation of NPM back into the nucleus. We also showed that NPM expression could be reduced by NPM siRNA. The knockdown efficiency of NPM siRNA is shown in Supplementary Figure S1.

Downregulation of NPM blocks anti-inflammatory effects of WA-25. (a, b) Western blot of iNOS. Relative densities of the bands are provided. *Significantly (P<0.05) different from the LPS group. #Significantly (P<0.05) different from the L+WA-25+siRNA group; n=6 per group. (c) Photos of animals from different groups on day 15. (d) AD scores, (e) TEWL levels, (f) scratching behavior and (g) alloknesis of the groups. (h) NPM immunostaining in the skin. Scale bar=8 μm. (i) LAMP-1 immunostaining in the skin. Scale bar=50 μm. *Significantly (P<0.05) different from the AD+W group; n=6 per group. TEWL, transepidermal water loss.
NPM-regulated WA-25 effects on AD
To clarify the therapeutic mechanisms by which WA-25 elicits its effects on AD and the role of NPM in these mechanisms, we used immunofluorescence and immunocytochemistry to further examine skin samples from animals in the various treatment groups. A recent study indicated that epidermal barrier dysfunction was the major manifestation of AD progression.32 As shown in Figure 5a, we used the expression of E-cadherin to assess the integrity of the epidermal barrier. These results showed that WA-25 restored AD-induced epidermal barrier dysfunction and that this restoration was blocked by NPM knockdown. As shown in Figure 5b, we found that AD actually leads to the downregulation of FLG-1 compared with the control group. Interestingly, WA-25 treatment was found to increase FLG-1 expression and NPM knockdown inhibited this WA-25-induced increase in FLG-1 expression. To determine how WA-25 relieves AD-induced itching or alloknesis, we performed immunocytochemistry to examine itch-related molecules. As shown in Figure 5c and d, AD caused the overexpression of both histamine and substance P in the skin, whereas WA-25 markedly reduced histamine and substance P expression. Similarly, NPM knockdown blocked the reduction of itch-related proteins. Because many AD patients develop chronic itching, recent research has indicated that astrocytes play a critical role in this process.33 Therefore, we used GFAP as an astrocyte marker to examine whether WA-25 could regulate astrocytes in the dorsal horn of the spinal cord. WA-25 was found to inhibit AD-stimulated astrocyte activation (Figure 5d), and NPM knockdown could disrupt this process. As AD also causes inflamed skin, we asked whether WA-25 could reduce AD-induced inflammation. Using an inflammation antibody array, we examined the expression levels of 40 inflammatory cytokines in the serum. Among the cytokines tested, WA-25 significantly inhibited the secretion of most inflammatory cytokines in the serum compared to the AD group (Supplementary Figure S2). Similarly, the data also showed that NPM knockdown disrupted the secretion of inflammatory cytokines that were modulated by WA-25.

NPM-regulated therapeutic effects of WA-25 on AD. (a) Immunostaining of E-cadherin (green) and DAPI (blue) in the skin. Scale bar=30 μm. (b) Immunostaining of FLG-1 (green) in the skin. Scale bar=100 μm. (c) Immunostaining of histamine and substance P in the skin. Scale bar=50 μm. (d) Immunostaining of glial fibrillary acidic protein (GFAP) (red) from the dorsal horn of the spinal cord. Scale bar=100 μm; n=6 per group.
Discussion
Our previous studies have indicated that WA-25 is effective in many inflammation-related diseases. However, whether WA-25 can be used as a treatment for AD remains unknown. We used a RAW 264.7 cell model to clarify the mechanisms by which WA-25 may elicit anti-inflammatory effects on AD. Our data suggest that WA-25 possesses anti-inflammatory and anti-AD activities. Furthermore, these observations led to the discovery that NPM may play an important role in regulating inflammation and AD.
Activated macrophages play a crucial role in inflammation.34 RAW 264.7 cells are a macrophage cell line that can induce inflammatory signals after LPS stimulation. Inflammation leads to the production of NO and prostaglandin E2, which are regulated by iNOS.35 Therefore, many anti-inflammatory drugs have been identified and developed by analyzing these target proteins.36 We found that WA-25 decreased the LPS-induced expression of iNOS. As expected, the upregulated levels of NO were also reduced by WA-25. These results mirror studies in which anti-inflammatory drugs were developed.37 Generally, substances that can regulate macrophage activation have good anti-inflammatory effects.38 However, high levels of ROS have been reported in many human diseases such as inflammation and cancer.39 Our observations suggest that WA-25 can reduce LPS-stimulated ROS production. In addition, we found that WA-25 upregulated the expression of both HO-1 and Nrf2. Recent studies have shown that HO-1 has anti-inflammatory, antioxidant and anti-apoptotic activities.40 Additionally, Nrf2 is a transcription factor that increases the expression of many genes encoding antioxidant enzymes,41 and WA-25 may prevent inflammation-induced oxidative stress. Given these results, we propose that WA-25 may serve as a potential anti-inflammatory drug that elicits anti-oxidative effects via the Nrf2-HO-1 signaling pathway.
Recent studies have indicated that AD is associated with inflammation, oxidative stress, skin barrier dysfunction and itching.42, 43, 44 Therefore, WA-25 may be an excellent therapy for use in patients with AD. Our results showed that WA-25 alleviated DNCB-induced AD to a greater extent than HC did (Figure 2b). Skin barrier defects in patients with AD are considered the primary phenotypes and can be measured by TEWL.45 As shown in Figure 2d, WA-25 inhibited AD-induced TEWL to a greater extent than HC. Itching is also a common symptom in early AD,46 and some AD cases develop alloknesis, which is characterized by extreme itching. Our results showed that WA-25 relieved AD-stimulated scratching behavior and alloknesis. Most importantly, the effects on scratching behavior and alloknesis were much greater than those of HC. As shown in Figure 2g, WA-25 was able to block inflammation by the regulation of iNOS. Together, the above data indicate that WA-25 has the potential to treat AD better than the currently used therapies.
Accumulating evidence has revealed that NF-κB is associated with several inflammatory diseases and plays an important role in transcription.47 When NF-κB is translocated into the nucleus, it activates numerous inflammatory signaling pathways. Drugs targeting NF-κB may prove to be successful therapies for treating inflammatory diseases.48 Our results demonstrate that WA-25 inhibits the translocation of NF-κB. Furthermore, WA-25 upregulates sirt-6 levels in the nucleus, which suppresses the transcription of NF-κB. A recent study reported that the overexpression of sirt-6 relieved arthritis by inhibiting anti-inflammatory responses.49 We propose that WA-25 can downregulate the ability of NF-κB to induce inflammation. However, the detailed mechanism involved in the regulation of NF-κB is still unclear. NPM is a phosphoprotein that shuttles between the nucleus and the cytoplasm.50 Generally, NPM is considered a regulator of cellular differentiation, proliferation and transformation and, as a result, may be associated with cancer.51 Hingorani et al.52 showed that NPM has multiple functions. However, the role of NPM in inflammation has not frequently been described. Our results suggest that WA-25 helps NPM translocate into the nucleus and bind NF-κB, thereby disrupting the NF-κB signaling pathway. As shown in Figure 3d, we found that high levels of NPM are colocalized with NF-κB after treatment with WA-25. Some studies reported that abnormal NPM levels accumulate in the cytoplasm.53, 54 Our results showed that NPM was almost exclusively found in the cytoplasm in the LPS group. Therefore, we propose that a mechanism exists by which abnormal NPM is degraded, thus preventing its entry into the nucleus. Lysosomes play a critical role in degrading organelles, proteins and other substances during homeostasis maintenance.55 We found that NPM might be degraded by lysosomes after its stimulation by LPS. However, few studies have demonstrated an interaction between NPM and lysosomes. We used immunoprecipitation assays to explore whether WA-25 actually induces NPM to bind NF-κB in the nucleus. From these results, we propose that WA-25 promotes NPM translocation into the nucleus, thereby inhibiting the LPS-induced NF-κB signaling pathway. In addition, we found that inflammation can cause NPM degradation by lysosomes in the cytoplasm. These data indicate that NPM shuttles between the cytoplasm and the nucleus, depending on the concurrent inflammatory state.
NSC 348884 is an NPM inhibitor that can induce cancer cell apoptosis.56 Therefore, we used NSC 348884 and NPM siRNA to examine whether the therapeutic effects of WA-25 are mediated by NPM. We observed that both NSC 348884 and NPM siRNA disrupted the downregulation of iNOS by WA-25. However, some studies reported that NPM may play a role in AD. Park et al.57 used two-dimensional electrophoresis to determine that NPM expression was altered in cases of AD. Interestingly, the effects of WA-25 on AD were disrupted by either NSC 348884 or NPM siRNA. Furthermore, in mice induced to develop AD, NPM was present in the cytoplasm of skin cells, similar to the results observed after the LPS stimulation of macrophages. We also found that NPM knockdown decreased the levels of the lysosomal marker LAMP1, suggesting the presence of fewer lysosomes in the cell. From these data, we propose that NPM can serve as a novel target for AD treatments, thereby reducing inflammation, skin barrier dysfunction and itching.
During AD progression, patients often experience skin barrier dysfunction.58 Therefore, some researchers believe that barrier-restoring therapies are extremely effective for AD.59 Because E-cadherin is responsible for cellular adhesion, Nakai et al.60 suggested that a reduced expression of E-cadherin would result in skin barrier dysfunction. From our results, we observed that WA-25 can recover AD-induced disruption of the skin barrier (Figure 5a). FLG also plays a major role in skin barrier dysfunction.61 Several studies have demonstrated that low FLG levels result in many skin diseases, such as atopic eczema and dry skin.62 Consistent with our results, we observed that WA-25 inhibited the AD-induced downregulation of FLG. Furthermore, this inhibition could be blocked by NPM knockdown. Thus, we suggest that WA-25 ameliorates skin barrier dysfunction via NPM.
Histamine and substance P have both been shown to play roles in AD-associated itching.63, 64 Therefore, antihistamines are often used to treat AD. As shown Figure 5c, we found that WA-25 could relieve AD-induced overexpression of both histamine and substance P. Therefore, it is apparent that WA-25 can inhibit itching. In addition, a recent study reported that substance P may cause chronic itching,65 which is considered a breakdown of itch-sensing neurons, and that it may cause the spinal cord to transmit abnormal signals to the brain.66 Additionally, alloknesis may be a central mechanism by which itch-related neurons in the spinal cord are activated.67 Therefore, astrocyte activation may cause alloknesis and chronic itching.29 Based on these hypotheses, we examined whether astrocytes were activated in the dorsal horn of the spinal cord. The data showed that WA-25 inhibited AD-induced astrocyte activation. Interestingly, NPM knockdown disrupted the anti-itching effects of WA-25. However, few studies have discussed an association between NPM and itching. Given these results, we proposed that NPM also plays an important role in chronic itching. Several studies have indicated that cytokines are involved in AD; therefore, therapies have been developed to modulate the expression of cytokines.68, 69 We used an inflammation antibody array to measure the expression levels of 40 inflammatory cytokines in the serum. We found that WA-25 inhibited the overexpression of nearly all AD-stimulated inflammatory cytokines. Furthermore, NPM knockdown blocked the downregulation of inflammatory cytokines by WA-25. Consistent with our previous results, WA-25 was shown to inhibit inflammation.
Our previous studies reported that WA-25 impedes the formation of foam cells, which would cause atherosclerosis by activating the transforming growth factor β1 (TGF-β1) pathway.24 Interestingly, TGF-β1 inhibits AD in NC/Nga mice.70 However, we also found that WA-25 inhibits angiogenesis.71 A recent study demonstrated that high levels of vascular endothelial growth factors-A (VEGF-A) are detectable in AD skin.72 Therefore, angiogenesis may play an important role in AD. We suggest that TGF-β1 or VEGF-A may be useful therapeutic targets for AD. However, a few reports have discussed the role of NPM in the TGF-β1 pathway or angiogenesis. Therefore, NPM may provide novel insight into TGF-β1- or VEGF-A-related diseases.
Taken together, we suggest that WA-25 is a better therapy for the treatment of AD than are the currently available therapies. Additionally, we found that NPM may play an important role in blocking inflammation and AD, which has not been discussed previously. However, the detailed mechanism by which NPM aids in the recovery of the skin barrier and in preventing itching should be investigated in the future.
References
Leung DY, Bieber T . Atopic dermatitis. Lancet 2003; 361: 151–160.
Gittler JK, Krueger JG, Guttman-Yassky E . Atopic dermatitis results in intrinsic barrier and immune abnormalities: implications for contact dermatitis. J Allergy Clin Immunol 2013; 131: 300–313.
Harper J, Ahmed I, Barclay G, Lacour M, Hoeger P, Cork M et al. Cyclosporin for severe childhood atopic dermatitis: short course versus continuous therapy. Br J Dermatol 2000; 142: 52–58.
Atherton DJ . Topical corticosteroids in atopic dermatitis: recent research reassures that they are safe and effective in the medium term. BMJ 2003; 327: 942.
Sulzberger MB . The effect of topically applied compound F in selected dermatoses. J Invest Dermatol 1952; 19: 101.
Rahman M, Nandi A, Kabir S, Kamal M, Basher M, Banu L . Topical Tacrolimus versus Hydrocortisone on atopic dermatitis in paediatric patients: a randomized controlled trial. Mymensingh Med J 2015; 24: 457–463.
Zirwas MJ, Barkovic S . Anti-pruritic efficacy of itch relief lotion and cream in patients with atopic history: comparison with hydrocortisone cream. J Drugs Dermatol 2017; 16: 243.
Homey B, Steinhoff M, Ruzicka T, Leung DY . Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol 2006; 118: 178–189.
Brown SJ, Asai Y, Cordell HJ, Campbell LE, Zhao Y, Liao H et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J Allergy Clin Immunol 2011; 127: 661–667.
Honda T, Egawa G, Grabbe S, Kabashima K . Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J Invest Dermatol 2013; 133: 303–315.
Kim TH, Kim GD, Jin YH, Park YS, Park CS . Omega-3 fatty acid-derived mediator, Resolvin E1, ameliorates 2, 4-dinitrofluorobenzene-induced atopic dermatitis in NC/Nga mice. Int Immunopharmacol 2012; 14: 384–391.
Mosser DM, Edwards JP . Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8: 958–969.
Mirzapoiazova T, Kolosova IA, Moreno L, Sammani S, Garcia JG, Verin AD . Suppression of endotoxin-induced inflammation by taxol. Eur Respir J 2007; 30: 429–435.
Otterbein LE, Choi AM . Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol 2000; 279: L1029–L1037.
Bach FH . Heme oxygenase-1: a therapeutic amplification funnel. FASEB J 2005; 19: 1216–1219.
Park CM, Park JY, Noh KH, Shin JH, Song YS . Taraxacum officinale Weber extracts inhibit LPS-induced oxidative stress and nitric oxide production via the NF-κB modulation in RAW 264.7 cells. J Ethnopharmacol 2011; 133: 834–842.
Xie Q, Kashiwabara Y, Nathan C . Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 1994; 269: 4705–4708.
Nawa Y, Kawahara K, Tancharoen S, Meng X, Sameshima H, Ito T et al. Nucleophosm in may act as an alarmin: implications for severe sepsis. J Leukoc Biol 2009; 86: 645–653.
Montaser R, Luesch H . Marine natural products: a new wave of drugs? Future Med Chem 2011; 3: 1475–1489.
Chen SC, Chien YC, Pan CH, Sheu JH, Chen CY, Wu CH . Inhibitory effect of dihydroaustrasulfone alcohol on the migration of human non-small cell lung carcinoma A549 cells and the antitumor effect on a Lewis lung carcinoma-bearing tumor model in C57BL/6J mice. Mar Drugs 2014; 12: 196–213.
Wen ZH, Chao CH, Wu MH, Sheu JH . A neuroprotective sulfone of marine origin and the in vivo anti-inflammatory activity of an analogue. Eur J Med Chem 2010; 45: 5998–6004.
Jean YH, Chen WF, Duh CY, Huang SY, Hsu CH, Lin CS et al. Inducible nitric oxide synthase and cyclooxygenase-2 participate in anti-inflammatory and analgesic effects of the natural marine compound lemnalol from Formosan soft coral Lemnalia cervicorni. Eur J Pharmacol 2008; 578: 323–331.
Wu GJ, Chen WF, Hung HC, Jean YH, Sung CS, Chakraborty C et al. Effects of propofol on proliferation and anti-apoptosis of neuroblastoma SH-SY5Y cell line: new insights into neuroprotection. Brain Res 2011; 1384: 42–50.
Wang YC, Hung HC, Feng CW, Huang SY, Chen CH, Lin YY et al. Dihydroaustrasulfone alcohol (WA-25) impedes macrophage foam cell formation by regulating the transforming growth factor-β1 pathway. Int J Mol Sci 2015; 16: 10507–10525.
Chan CC, Liou CJ, Xu PY, Shen JJ, Kuo ML, Len WB et al. Effect of dehydroepiandrosterone on atopic dermatitis-like skin lesions induced by 1-chloro-2, 4-dinitrobenzene in mouse. J Dermatol Sci 2013; 72: 149–157.
Sun J, Zhao Y, Hu J . Curcumin inhibits imiquimod-induced psoriasis-like inflammation by inhibiting IL-1beta and IL-6 production in mice. PLoS ONE 2013; 8: e67078.
Karki R, Jung MA, Kim KJ, Kim DW . Inhibitory effect of Nelumbo nucifera (Gaertn.) on the development of atopic dermatitis-like skin lesions in NC/Nga mice. Evid Based Complement Alternat Med 2012; 2012: 153568.
Ryu KR, Choi JY, Chung S, Kim DH . Anti-scratching behavioral effect of the essential oil and phytol isolated from Artemisia princeps Pamp. in mice. Planta Med 2011; 77: 22–26.
Liu T, Han Q, Chen G, Huang Y, Zhao LX, Berta T et al. Toll-like receptor 4 contributes to chronic itch, alloknesis, and spinal astrocyte activation in male mice. Pain 2016; 157: 806–817.
Hegde V, Hickerson RP, Nainamalai S, Campbell PA, Smith FJ, McLean WI et al. In vivo gene silencing following non-invasive siRNA delivery into the skin using a novel topical formulation. J Control Release 2014; 196: 355–362.
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 2009; 136: 62–74.
Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol 2009; 129: 1892–1908.
Green D, Dong X . Supporting itch: a new role for astrocytes in chronic itch. Nat Med 2015; 21: 841.
Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol 2005; 142: 481–489.
Dahiya Y, Pandey RK, Bhatt KH, Sodhi A . Role of prostaglandin E2 in peptidoglycan mediated iNOS expression in mouse peritoneal macrophages in vitro. FEBS Lett 2010; 584: 4227–4232.
Bertolini A, Ottani A, Sandrini M . Selective COX-2 inhibitors and dual acting anti-inflammatory drugs: critical remarks. Curr Med Chem 2002; 9: 1033–1043.
Kanno Si Kakuta M, Kitajima Y, Osanai Y, Kurauchi K, Ohtake T et al. Inhibitory effect of trimidox on lipopolysaccharide-induced nitric oxide production in RAW 264.7 macrophages. J Pharmacol Sci 2007; 104: 278–281.
Tripathi S, Bruch D, Kittur DS . Ginger extract inhibits LPS induced macrophage activation and function. BMC Complement Altern Med 2008; 8: 1.
Naik E, Dixit VM . Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 2011; 208: 417–420.
Ryter SW, Alam J, Choi AM . Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006; 86: 583–650.
Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y et al. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12, 14-prostaglandin J2. Mol Cell Biol 2004; 24: 36–45.
Agrawal R, Woodfolk JA . Skin barrier defects in atopic dermatitis. Curr Allergy Asthma Rep 2014; 14: 1–11.
Omata N, Tsukahara H, Ito S, Ohshima Y, Yasutomi M, Yamada A et al. Increased oxidative stress in childhood atopic dermatitis. Life Sci 2001; 69: 223–228.
Yarbrough KB, Neuhaus KJ, Simpson EL . The effects of treatment on itch in atopic dermatitis. Dermatol Ther 2013; 26: 110–119.
Kelleher M, Dunn-Galvin A, Hourihane JOB, Murray D, Campbell LE, McLean WI et al. Skin barrier dysfunction measured by transepidermal water loss at 2 days and 2 months predates and predicts atopic dermatitis at 1 year. J Allergy Clin Immunol 2015; 135: e1.
Inagaki N, Igeta K, Kim JF, Nagao M, Shiraishi N, Nakamura N et al. Involvement of unique mechanisms in the induction of scratching behavior in BALB/c mice by compound 48/80. Eur J Pharmacol 2002; 448: 175–183.
Tak PP, Firestein GS . NF-kappaB: a key role in inflammatory diseases. J Clin Invest 2001; 107: 7–11.
Ben-Neriah Y, Karin M . Inflammation meets cancer, with NF-[kappa] B as the matchmaker. Nat Immunol 2011; 12: 715–723.
Lee HS, Ka SO, Lee SM, Lee SI, Park JW, Park BH . Overexpression of Sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheum 2013; 65: 1776–1785.
Borer R, Lehner C, Eppenberger H, Nigg E . Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989; 56: 379–390.
Grisendi S, Mecucci C, Falini B, Pandolfi PP . Nucleophosmin and cancer. Nat Rev Cancer 2006; 6: 493–505.
Hingorani K, Szebeni A, Olson MO . Mapping the functional domains of nucleolar protein B23. J Biol Chem 2000; 275: 24451–24457.
Falini B, Bolli N, Liso A, Martelli M, Mannucci R, Pileri S et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 2009; 23: 1731–1743.
Luo J, Qi C, Xu W, Kamel-Reid S, Brandwein J, Chang H . Cytoplasmic expression of nucleophosmin accurately predicts mutation in the nucleophosmin gene in patients with acute myeloid leukemia and normal karyotype. Am J Clin Pathol 2010; 133: 34–40.
Desnick RJ, Schuchman EH . Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet 2002; 3: 954–966.
Qi W, Shakalya K, Stejskal A, Goldman A, Beeck S, Cooke L et al. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene 2008; 27: 4210–4220.
Park YD, Lyou YJ, Yang JM . Two-dimensional electrophoresis analyses of atopic dermatitis and the chances to detect new candidate proteins by the variations in immobilized pH gradient strips. J Dermatol Sci 2007; 47: 9–17.
Marsella R, Olivry T, Carlotti DN . Current evidence of skin barrier dysfunction in human and canine atopic dermatitis. Vet Dermatol 2011; 22: 239–248.
Valdman-Grinshpoun Y, Ben-Amitai D, Zvulunov A . Barrier-restoring therapies in atopic dermatitis: current approaches and future perspectives. Dermatol Res Pract 2012; 2012: 923134.
Nakai K, Yoneda K, Hosokawa Y, Moriue T, Presland RB, Fallon PG et al. Reduced expression of epidermal growth factor receptor, E-cadherin, and occludin in the skin of flaky tail mice is due to filaggrin and loricrin deficiencies. Am J Pathol 2012; 181: 969–977.
Armengot-Carbo M, Hernández-Martín A, Torrelo A . The role of filaggrin in the skin barrier and disease development. Actas Dermosifiliogr 2015; 106: 86–95.
Sandilands A, Sutherland C, Irvine AD, McLean WI . Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci 2009; 122: 1285–1294.
Anand P, Springall D, Blank M, Sellu D, Polak J, Bloom S . Neuropeptides in skin disease: increased VIP in eczema and psoriasis but not axillary hyperhidrosis. Br J Dermatol 1991; 124: 547–549.
Williams DH . Skin temperature reaction to histamine in atopic dermatitis (disseminated neurodermatitis). J Invest Dermatol 1938; 1: 119–129.
Potenzieri C, Undem BJ . Basic mechanisms of itch. Clin Exp Allergy 2012; 42: 8–19.
Oaklander AL . Neuropathic itch. Semin Cutan Med Surg 2011; 30: 87–92.
Akiyama T, Carstens MI, Ikoma A, Cevikbas F, Steinhoff M, Carstens E . Mouse model of touch-evoked itch (alloknesis). J Invest Dermatol 2012; 132: 1886–1891.
Auriemma M, Vianale G, Amerio P, Reale M . Cytokines and T cells in atopic dermatitis. Eur Cytokine Netw 2013; 24: 37–44.
Noda S, Krueger JG, Guttman-Yassky E . The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol 2015; 135: 324–336.
Sumiyoshi K, Nakao A, Ushio H, Mitsuishi K, Okumura K, Tsuboi R et al. Transforming growth factor-bβ1 suppresses atopic dermatitis-like skin lesions in NC/Nga mice. Clin Exp Allergy 2002; 32: 309–314.
Lin SW, Huang SC, Kuo HM, Chen CH, Ma YL, Chu TH et al. Coral-derived compound WA-25 inhibits angiogenesis by attenuating the VEGF/VEGFR2 signaling pathway. Mar Drugs 2015; 13: 861–878.
Genovese A, Detoraki A, Granata F, Galdiero M, Spadaro G, Marone G . Angiogenesis, lymphangiogenesis and atopic dermatitis. Chem Immunol Allergy 2012; 96: 50–60.
Acknowledgements
The study was partly supported by grants from the Ministry of Science and Technology, Taiwan (103-2628-B-110-002-MY3 and 105-2325-B-110-001) and Taipei Veterans General Hospital (V105C-109).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hung, HC., Feng, CW., Lin, YY. et al. Nucleophosmin modulates the alleviation of atopic dermatitis caused by the marine-derived compound dihydroaustrasulfone alcohol. Exp Mol Med 50, e446 (2018). https://doi.org/10.1038/emm.2017.272
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.272
