
Abstract
In order to ensure normal body function, the human body is dependent on a tight control of its blood glucose levels. This is accomplished by a highly sophisticated network of various hormones and neuropeptides released mainly from the brain, pancreas, liver, intestine as well as adipose and muscle tissue. Within this network, the pancreas represents a key player by secreting the blood sugar-lowering hormone insulin and its opponent glucagon. However, disturbances in the interplay of the hormones and peptides involved may lead to metabolic disorders such as type 2 diabetes mellitus (T2DM) whose prevalence, comorbidities and medical costs take on a dramatic scale. Therefore, it is of utmost importance to uncover and understand the mechanisms underlying the various interactions to improve existing anti-diabetic therapies and drugs on the one hand and to develop new therapeutic approaches on the other. This review summarizes the interplay of the pancreas with various other organs and tissues that maintain glucose homeostasis. Furthermore, anti-diabetic drugs and their impact on signaling pathways underlying the network will be discussed.
Similar content being viewed by others

The pancreas is an exocrine and endocrine organ
The pancreas has key roles in the regulation of macronutrient digestion and hence metabolism/energy homeostasis by releasing various digestive enzymes and pancreatic hormones. It is located behind the stomach within the left upper abdominal cavity and is partitioned into head, body and tail. The majority of this secretory organ consists of acinar—or exocrine—cells that secrete the pancreatic juice containing digestive enzymes, such as amylase, pancreatic lipase and trypsinogen, into the ducts, that is, the main pancreatic and the accessory pancreatic duct. In contrast, pancreatic hormones are released in an endocrine manner, that is, direct secretion into the blood stream. The endocrine cells are clustered together, thereby forming the so-called islets of Langerhans, which are small, island-like structures within the exocrine pancreatic tissue that account for only 1–2% of the entire organ (Figure 1).1 There are five different cell types releasing various hormones from the endocrine system: glucagon-producing α-cells,2 which represent 15–20% of the total islet cells; amylin-, C-peptide- and insulin-producing β-cells,2 which account for 65–80% of the total cells; pancreatic polypeptide (PP)-producing γ-cells,3 which comprise 3–5% of the total islet cells; somatostatin-producing δ-cells,2 which constitute 3–10% of the total cells; and ghrelin-producing ɛ-cells,4 which comprise <1% of the total islet cells. Each of the hormones has distinct functions. Glucagon increases blood glucose levels, whereas insulin decreases them.5 Somatostatin inhibits both, glucagon and insulin release,6 whereas PP regulates the exocrine and endocrine secretion activity of the pancreas.3, 7 Altogether, these hormones regulate glucose homeostasis in vertebrates, as described in more detail below. Although the islets have a similar cellular composition among different species, that is, human, rat and mouse, their cytoarchitecture differs greatly. Although islets in rodents are primarily composed of β-cells located in the center with other cell types in the periphery, human islets exhibit interconnected α- and β-cells.2, 8

Anatomical organization of the pancreas. The exocrine function of the pancreas is mediated by acinar cells that secrete digestive enzymes into the upper small intestine via the pancreatic duct. Its endocrine function involves the secretion of various hormones from different cell types within the pancreatic islets of Langerhans. The micrograph shows the pancreatic islets. LM × 760 (Micrograph provided by the Regents of University of Michigan Medical School © 2012). Adapted from Human Anatomy and Physiology, an OpenStax College resource.404
Through its various hormones, particularly glucagon and insulin, the pancreas maintains blood glucose levels within a very narrow range of 4–6 mM. This preservation is accomplished by the opposing and balanced actions of glucagon and insulin, referred to as glucose homeostasis. During sleep or in between meals, when blood glucose levels are low, glucagon is released from α-cells to promote hepatic glycogenolysis. In addition, glucagon drives hepatic and renal gluconeogenesis to increase endogenous blood glucose levels9 during prolonged fasting. In contrast, insulin secretion from β-cells is stimulated by elevated exogenous glucose levels, such as those occurring after a meal.10 After docking to its receptor on muscle and adipose tissue, insulin enables the insulin-dependent uptake of glucose into these tissues and hence lowers blood glucose levels by removing the exogenous glucose from the blood stream (Figure 2).11, 12, 13 Furthermore, insulin promotes glycogenesis,14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26 lipogenesis27, 28 and the incorporation of amino acids into proteins;29 thus, it is an anabolic hormone, in contrast to the catabolic activity of glucagon.

Maintenance of blood glucose levels by glucagon and insulin. When blood glucose levels are low, the pancreas secretes glucagon, which increases endogenous blood glucose levels through glycogenolysis. After a meal, when exogenous blood glucose levels are high, insulin is released to trigger glucose uptake into insulin-dependent muscle and adipose tissues as well as to promote glycogenesis.
The insulin secretion signaling pathway
Endocrine cells secrete their respective hormones in response to external signals, such as nutrient intake or stress, via humoral, neural or hormonal signaling pathways. The underlying molecular process that translates the stimulus into the actual hormone release is called stimulus-secretion coupling which is known as the stimulus-dependent exocytosis of a particular substance, such as glucose-stimulated β-cell insulin release.30
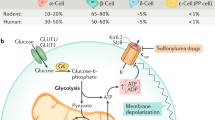
In β-cells, the main stimulus for insulin release are elevated blood glucose levels following a meal.10 The circulating blood glucose is taken up by the facilitative glucose transporter GLUT2 (SLC2A2), which is located on the surface of the β-cells. Once inside the cell, glucose undergoes glycolysis, thereby generating adenosine triphosphate (ATP), resulting in an increased ATP/ADP ratio. This altered ratio then leads to the closure of ATP-sensitive K+-channels (KATP-channels). Under non-stimulated conditions, these channels are open to ensure the maintenance of the resting potential by transporting positively charged K+-ions down their concentration gradient out of the cell. Upon closure, the subsequent decrease in the magnitude of the outwardly directed K+-current elicits the depolarization of the membrane, followed by the opening of voltage-dependent Ca+-channels (VDCCs). The increase in intracellular calcium concentrations eventually triggers the fusion of insulin-containing granules with the membrane and the subsequent release of their content.31 The whole secretory process is biphasic with the first phase peaking around 5 minutes after the glucose stimulus with the majority of insulin being released during this first phase. In the second, somewhat slower, phase, the remaining insulin is secreted.32, 33, 34 Insulin is stored in large dense-core vesicles that are recruited to the proximity of the plasma membrane following stimulation such that insulin is readily available.35, 36 The key molecules that mediate the fusion of the insulin-containing large dense-core vesicles are the synaptosomal-associated protein of 25 kDa (SNAP-25), syntaxin-1 and synaptobrevin 2 (or vesicle-associated membrane protein VAMP2), all of which belong to the superfamily of the soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor proteins (SNAREs). Together with the Sec1/Munc18-like (SM) proteins they form the so-called SNARE complex.37 To initiate fusion, synaptobrevin 2, a vesicle (v-)SNARE that is integrated into the vesicle’s membrane, fuses with the target (t-)SNAREs syntaxin-1 and SNAP-25, which are located in the target cell membrane,38, 39 with mammalian uncoordinated (munc)-18 playing a key regulatory role (Figure 3).40, 41

Glucose-stimulated insulin release from a pancreatic β-cell. Exogenous glucose is taken up by GLUT2 and undergoes glycolysis inside the cell. Elevated adenosine triphosphate (ATP) levels alter the ATP/ADP ratio, which in turn leads to the closure of ATP-sensitive K+-channels. The subsequent membrane depolarization opens voltage-dependent Ca2+-channels in response to increasing intracellular calcium levels, which eventually trigger insulin secretion following vesicle fusion with the membrane.
To date, numerous SNARE isoforms, including syntaxin-1, -3 and -4, SNAP-25 and -23, as well as syntaptobrevins 2 and 3 (VAMP2 and 3), have been shown to be involved in glucose-stimulated insulin secretion,42, 43, 44, 45, 46 whereas VAMP8, a non-essential SNARE protein for glucose-stimulated insulin secretion, has a role in the regulation of the glucagon-like peptide-1-potentiated insulin secretion.47 In addition to SNARE and SM proteins, a calcium sensor is required for the initiation of membrane fusion. Synaptotagmins, which are highly expressed in neurons and endocrine cells, were shown to participate in Ca2+-dependent exocytosis processes. To date, 17 synaptotagmins (Syts 1–17) have been identified and only eight of them, namely Syt-1, -2, -3, -5, -6, -7, -9 and -10, are able to bind calcium.48 Following Ca2+-binding, synaptotagmins form a complex with the SNAREs to facilitate and trigger the vesicle-membrane fusion process. Among the synaptotagmin family, Syt-3, -5, -7, -8 and -9 are implicated in insulin exocytosis.49, 50, 51, 52
External factors affecting pancreatic hormone secretion
Metabolism–cAMP coupling
The glucose-triggered stimulus-secretion coupling is an established paradigm of insulin secretion from β-cells and includes a great variety of modulators that trigger, potentiate or inhibit glucose-stimulated insulin secretion, primarily through G-protein-coupled receptors (GPCRs). The most traditional external factor that initiates insulin secretion is glucose. In addition to its trigger function, glucose also induces pathways that amplify insulin secretion through metabolism-cAMP (cyclic adenosine monophosphate) coupling or the incretin hormones glucagon-like peptide (GLP)-1 and glucose-dependent insulinotropic peptide (GIP).31 Metabolism–cAMP coupling refers to the signaling cascade that occurs after the conversion of ATP, which is generated during intracellular glucose metabolism, into cAMP by adenylate cyclase (AC),53 which in turn activates protein kinase A (PKA)54 and cAMP-regulated guanine nucleotide exchange factors, also referred to as exchange protein directly activated by cAMP (Epac) 2.55, 56 Although Epac2 activation amplifies insulin secretion by mobilizing calcium from internal stores to increase Ca2+ levels57, 58 and by controlling the granule density in proximity to the plasma membrane,59 activated PKA exerts its effects by modulating KATP-channel60, 61 and calcium channel62, 63 activity through phosphorylation, thereby enhancing the number of highly Ca2+-sensitive insulin-containing granules64 and the probability of releasing secretory vesicles from the readily releasable pool,65 respectively.
The incretins GLP-1 and GIP
The gut-derived hormones GLP-1 and GIP, which are secreted from enteroendocrine L-cells66 and K-cells,67 respectively, upon glucose,66, 68 fructose,69 amino acid70 and free fatty acid (FFA)71, 72 ingestion, also potentiate insulin release through the so-called incretin effect. This effect describes the observation that orally, but not intravenously, administered glucose enhances insulin secretion by triggering GLP-1 and GIP secretion;73, 74, 75 the resulting potentiation of insulin secretion may account for up to 50% of the total release. The underlying mechanism includes GLP-1 and GIP binding to their GPCRs (GLP-1R and GIPR), both of which are expressed in pancreatic β-cells.76 The binding induces a conformational change in the receptors’ structure, followed by the exchange of guanosine diphosphate for guanosine triphosphate and the subsequent dissociation of the Gsα-subunit from the receptors. This subunit, in turn, activates adenylate cyclase to convert ATP into cAMP, thereby stimulating the cAMP signaling pathway described above.77, 78, 79, 80, 81, 82 Furthermore, GLP-1 increases intracellular calcium concentrations by mobilizing Ca2+ from ryanodine-sensitive stores83, 84 or, similar to GIP, by acting on voltage-dependent Ca2+-channels,85 thereby potentiating insulin release.85, 86, 87 Recent studies have also shown that GLP-1R agonists, such as exendin-488, induce the PKA-mediated phosphorylation of Snapin or Synaptotagmin-7, which in turn enhances GSIS by Snapin interacting with SNAP-2589 or by directly enhancing glucose- and Ca2+-triggered insulin release.90
Free Fatty Acids
FFAs not only stimulate incretin secretion but are also known to modulate insulin release through fatty acid metabolism. Although long-chain FFAs augment insulin secretion, short-chain FFAs inhibit it. The binding and subsequent interaction of long-chain FFAs with the G-protein-coupled free fatty acid receptor (FFAR) 1 in the pancreatic β-cells leads to the activation of phospholipase C. PLC then hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to diacylglycerol and inositol-1,4,5-triphosphate (IP3), with the latter docking on a calcium channel in the endoplasmic reticulum. The subsequent release of Ca2+ into the cytosol increases the intracellular Ca2+ concentration, which eventually triggers insulin secretion.91, 92, 93, 94 In contrast, short-chain FFAs inhibit glucose-stimulated insulin secretion due to decreased glucose oxidation and the subsequently decreased ATP/ADP ratio.95 Another inhibitor of insulin release is stress, specifically norepinephrine (noradrenaline) produced in response to stress.96 Norepinephrine binds to its α2-adrenergic receptors, which are linked to GPCRs, resulting in the inhibition of AC as well as in hyperpolarization. This prevents an increase in the cytosolic Ca2+ concentration and, subsequently, insulin secretion.97, 98
Interplay between the pancreatic islets and other organs
The brain–islet axis
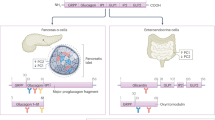
Just as insulin exerts its effects on other organs and tissues, other organs interact with the pancreas to modulate insulin secretion (Figure 4). One of these interacting organs is the brain, which comprises the mutual brain–islet axis that interacts with the pancreas and vice versa. The pancreas is highly innervated with both, parasympathetic99, 100 and sympathetic100, 101 nerve fibers from the autonomic nervous system. At the same time, insulin receptors are widely distributed within the brain, including the hypothalamus, cerebral cortex, cerebellum102 and hippocampal formation103 in humans, as well as the olfactory and limbic areas,104, 105 hypothalamus106—particularly the periventricular nucleus107 and the arcuate nucleus108, 109—hippocampus and the choroid plexus105 in rat brains. Lesions in various brain regions were shown to affect pancreatic hormone secretion. The destruction of the ventromedial hypothalamus results not only in insulin hypersecretion110, 111, 112 due to loss of the ventromedial hypothalamus-mediated inhibitory impact on pancreatic β-cells113 but also in higher glucagon levels.111, 112 Glucagon secretion may also be modulated by the hypothalamic brain-derived neurotrophic factor114 via efferent nerves,115 whereas the melanocortin system directly reduces basal insulin levels by stimulating sympathetic nerve fibers via α-adrenoceptors.116 Acting via α-adrenoceptors,117 norepinephrine also inhibits insulin secretion,96 which is an important aspect of the fight-or-flight response. The neurotransmitter Neuropeptide Y (NPY), which is mainly expressed in the sympathetic nerve fibers of the autonomic nervous system, also blunts insulin release,118, 119 and the loss of NPY’s inhibitory action results in elevated basal and glucose-stimulated insulin secretion as well as in increased islet mass.120 NPY binding to its GPCR Y1 causes the activated Giα-subunit to block adenylate cyclase activation, which in turn inhibits the cAMP pathway.121 Furthermore, the NPY-mediated inhibition was shown to be Gβγ- and Ca2+-independent.122 In addition to the well-known insulin stimulator acetylcholine, which exerts its effects via M3 muscarinic receptors,123 melanin concentrating hormone, vasoactive intestinal peptide (VIP), its close relative pituitary adenylate cyclase-activating polypeptide (PACAP) and gastrin-releasing peptide also promote insulin and, in the case of VIP124 and PACAP,125 glucagon release. The various neuropeptides exert their effects through various pathways, including the extracellular signal-regulated kinase (ERK)/Akt pathway, and modulation of Ca2+-influx (melanin concentrating hormone),126 cAMP and, to a lesser extent, PI3K signaling (VIP and PACAP),127, 128 muscarinic/β-adrenoceptors signaling, PI3K/PKC signaling and Ca2+-mobilization from intracellular stores (gastrin-releasing peptide).129, 130

The interplay of the pancreas with the brain, liver, gut as well as adipose and muscle tissue. The pancreas interacts with the brain, liver, gut and adipose and muscle tissue in a highly sophisticated network via various hormones, neurotransmitters and cytokines. BNDF, brain-derived neurotrophic factor; CCK, cholecystokinin; GIP, glucose-dependent insulinotropic peptide; GLP-1, glucagon-like peptide 1; GRP, gastrin-releasing peptide; IL-6, Interleukin 6; MCH, melanin concentrating hormone; NPY, neuropeptide Y; PACAP, pituitary adenylate cyclase-activating polypeptide; POMC, pro-opiomelanocortin; VIP, vasoactive intestinal peptide.
Likewise, insulin release is stimulated by the so-called cephalic phase, which represents the conditioned reflex of increased hormone secretion, referred to as cephalic phase insulin response,131 even in the absence of nutrients/glucose as a trigger,132, 133, 134 such as when anticipating a meal, to prepare the organism to adequately respond to incoming nutrients.135 Moreover, cephalic phase insulin response is pivotal for ensuring normal postprandial glucose management.136 The neural mechanism underlying cephalic phase insulin response was found to include cholinergic and non-cholinergic processes136 as well as the dorsal vagal complex located in the medulla oblongata.137 Conversely, insulin released in response to a meal enters the brain via the blood–brain–barrier138 to decrease food intake139, 140 by stimulating hypothalamic pro-opiomelanocortin neurons141 and initiating the PI3K signaling pathway142 in these pro-opiomelanocortin neurons.143 In contrast to its pro-opiomelanocortin-stimulating action, insulin inhibits NPY expression144 in Agouti-related peptide (AgRP/NPY) neurons, which are known to secrete the orexigenic neuropeptides NPY145, 146, 147 and AgRP.148, 149 Both, peripheral and central insulin signaling are impaired in obese or diabetic states.150, 151, 152, 153, 154
The liver–islet axis
The second group represents the liver–islet axis. The liver has a key role in glucose homeostasis by storing (glycogenesis) or releasing (glycogenolysis/gluconeogenesis) glucose upon interaction with insulin and glucagon, respectively. The binding of glucagon to its hepatic GPCR evokes the signaling cascade described under ‘External factors affecting pancreatic hormone secretion’, eventually resulting in the activation of PKA, which in turn stimulates two processes; one promotes glycogenolysis/gluconeogenesis and the other inhibits glycolysis/glycogenesis.155, 156 Glycogenolysis is a multistep process that includes the PKA-mediated phosphorylation of phosphorylase kinase,157 cleavage of glucose-1-phosphate (G-1-P) from glycogen by activated glycogen phosphorylase a158 and the conversion of G-1-P into G-6-P,159 eventually resulting in phosphate and free glucose. Hepatic gluconeogenesis is promoted by the PKA-mediated phosphorylation of the cAMP response element-binding protein, which in turn upregulates peroxisome proliferator-activated receptor-γ coactivator (PGC)-1.160 Together with the hepatocyte nuclear factor (HNF)-4, PGC-1 induces the transcription of phosphoenolpyruvate carboxykinase,161 which catalyzes the conversion of oxaloacetate into phosphoenolpyruvate, a rate-limiting step in gluconeogenesis. This is followed by reversed glycolysis, during which stimulation of the bifunctional PFK-2/FBPase-2 leads to both, enhanced gluconeogenesis through the abrogation of disabled fructose-1,6-bisphosphatase (FBPase)-1, which facilitates the successive conversion of substrates into G-6-P, and to suppressed glycolysis.162, 163 Glycolysis is further inhibited by the PKA-mediated inactivation of pyruvate kinase,164, 165, 166 resulting in the production of glucose instead of pyruvate. In addition, glucagon was found to suppress pyruvate kinase gene expression as well as to enhance pyruvate kinase mRNA degradation.167, 168 Finally, the PKA-induced inactivation of hepatic glycogen synthase169, 170, 171 decreases glycogen synthesis and concomitantly increases the hepatic glucose pool.
As glucagon’s opponent, insulin stimulates glycolysis via enhanced expression of the hepatic glucokinase gene,14, 15 a key enzyme that converts glucose into G-6-P. This increase is mediated by the sterol regulatory element binding protein-1c15 and requires the absence of cAMP.14 Furthermore, insulin inactivates glycogen phosphorylase and glycogen synthase kinase (GSK)-3172 through the PI3K pathway, which in turn activates glycogen synthase.18, 19, 20 The second liver-specific effect of insulin is to repress the expression of the phosphoenolpyruvate carboxykinase and G-6-Pase genes; the first by disrupting the association of cAMP response element-binding protein and RNA polymerase II with the phosphoenolpyruvate carboxykinase gene promoter,23 whereas G-6-P suppression requires PKBα/Akt and forkhead transcription factor (FOXO1),24, 25 whose expression was shown to be diminished by the inhibition of GSK-3.26
It is not only insulin and glucagon acting on the liver; hepatocyte-derived factors conversely influence the pancreas and/or insulin secretion. Although HNF3β was proposed to be pivotal for the transcription of the pancreatic and duodenal homeobox 1 (pdx1 or insulin promotor factor 1 (IPF-1)) gene, a transcription factor regulating pancreatic development173, 174175, it is the loss of HNF1α resulting in an almost abolished insulin secretion, likely due to a decreased response to intracellular calcium. These findings support the importance of HNF1α in maintaining β-cell function176 and its involvement in maturity-onset diabetes of the young (MODY3).177
The hepatokine betatrophin, also known as TD26, re-feeding induced fat and liver (RIFL), lipasin or angiopoietin-like (ANGPTL) 8, was first identified as a factor that drives β-cell proliferation and thus increases β-cell mass in a murine model of insulin resistance.178 Subsequent studies, however, did not reveal impairments in glucose homeostasis179 or β-cell expansion in Angptl8 knockout mice.180 Moreover, betatrophin does not have an effect on human β-cell replication, challenging its usefulness in diabetes therapy.181 This is substantiated by the fact that betatrophin levels are higher in T2DM patients,182, 183, 184 although they were lower in one study.185 However, this is likely to be due to technical issues.186
The gut–islet axis
Another important axis is the gut–islet axis. The gut releases various hormones upon nutrient ingestion, including GLP-1 and GIP, that bind to their respective receptors on pancreatic β-cells to potentiate insulin secretion, as described under ‘External factors affecting pancreatic hormone secretion’. Furthermore, both hormones exert pancreatic effects, such as GLP-1-stimulated insulin gene expression,77, 187 incretin-induced β-cell neogenesis, proliferation188, 189, 190, 191 and survival,192 the prevention of β-cell apoptosis in general193, 194 and in response to glucolipotoxicity.195 The extrapancreatic actions of GLP-1 include suppression of endogenous glucose production196/glycogenolysis,197 glucagon secretion,197, 198 appetite,199, 200 a delay in gastric emptying198, 199 and improved β-cell insulin sensitivity199, 201, 202 and glucose disposal,203, 204 whereas GIP positively affects lipid205, 206, 207 and bone metabolism.208, 209, 210, 211 Thus, GLP-1 and GIP mediate insulin secretion and concomitantly, insulin modulates GIP212 and GLP-1 release; the latter ocurring through the PI3K/Akt- and mitogen-activated protein kinase kinase (MAPKK or MEK)/ERK1/2 pathway.213 The importance of this interplay is also demonstrated by defective insulin responses and consequent glucose intolerance in GLP-1R−/− and GIPR−/− mice214, 215, 216, 217, 218 as well as in the pathogenesis of T2DM.219, 220, 221, 222, 223
In addition to incretins, there are the so-called decretins, namely limostatin and Neuromedin U (NmU), which are secreted during fasting to suppress insulin release. NmU, a (neuro)peptide that mediates the contraction of smooth muscles in the uterus (hence the ‘U’) among others, was first isolated from the pig spinal cord.224 Further mRNA expression studies, however, revealed NmU to be highly expressed in the gastrointestinal (GI) tract with the highest levels found in the upper GI, that is, duodenum and jejunum.225, 226 Within the GI structure, NmU is mainly located in submucosal and myenteric cells,227, 228 indicating its possible involvement in the neuronal control of GI function.229 In addition to this, NmU is likely to regulate insulin secretion; the G-protein-coupled NmU receptor 1 (NmUR1) is expressed in pancreatic islets and its simulation dose dependently decreased insulin release.230, 231 The underlying mechanism involves the simultaneous release of somatostatin—a known modulator of insulin secretion6—upon NmUR1 activation.232 A very recent study showed231 that the peptide hormone limostatin, which is expressed in Drosophila melanogaster, also reduces insulin secretion and its absence caused hyperinsulinemia, hypoglycemia and obesity. Moreover, knockdown of the fly NmUR orthologue not only reproduced the consequences of limostatin deficiency but also diminished its insulin-suppressing ability. Limostatin release is initiated by food depletion and hence may represent a novel mechanism for modulating insulin secretion during fasting.
Other gastrointestinal hormones that interact with the pancreas are gastrin and cholecystokinin (CCK). Gastrin, which is secreted from G-cells in the stomach and duodenum, acts as an islet growth factor, together with transforming growth factor-α, by promoting differentiation of ductular precursor cells233 and β-cell neogenesis as well as by enhancing the islet mass from transdifferentiated exocrine pancreatic tissue.234 Furthermore, it induces the expression of glucagon genes in α-cells.235 Along the same lines, CCK, which is synthesized and released from duodenal I-cells, potentiates basal, glucose-236, 237 and amino acid-induced insulin secretion,238 and augments glucagon secretion.237, 239 The pivotal role of CCK in modulating glucose homeostasis is reflected in postprandial hyperglycemia, which is due to reduced CCK plasma levels in noninsulin-dependent diabetes mellitus.240
Another important factor that is related to metabolic disorders such as obesity, T2DM and type 1 DM (T1DM) is the gut microbiota. Obesity, T2DM and T1DM patients display alterations in the composition of their microbiota that may initiate and/or promote the respective disorder. Recent findings linked an aberrant microbiome, which is generally represented by diminished diversity, including fewer butyrate-producing (butyrate was shown to trigger mucin production and hence gut integrity) and mucin-degrading bacteria,241 to the development of autoimmunity in T1DM.242 An altered microbiota composition may also contribute to obesity243, 244 as well as to T2DM245, 246, 247 and ‘correction’ by antibiotics,248 probiotics249 or prebiotics, the last of which causing a short-chain FFA-stimulated increase in GLP-1,250 may improve the disease condition.251
The adipocytes/myocytes–islet axis
On one hand, insulin’s interplay with adipose and muscle tissues is broadly based on facilitating insulin-dependent glucose uptake through the glucose transporter 4 (GLUT4).11, 12, 13 On the other hand, adipokines and myokines secreted from the adipose and muscle tissue, respectively, modulate insulin release. As part of the so-called adipoinsular axis,252 leptin, the most famous adipokine, mainly acts on its receptors in the hypothalamic arcuate nucleus to inhibit food intake and control whole body homeostasis.253 However, leptin receptor (Ob-R) mRNA expression was also observed in pancreatic islets254 and its stimulation caused a reduction in insulin secretion255, 256, 257 due to the activation of KATP-channels, which in turn prevented Ca2+-influx258 and the subsequent signaling pathway. Furthermore, leptin was shown to suppress insulin gene expression,259, 260 representing a negative feedback loop. Conversely, insulin enhances ob gene expression and leptin secretion.261, 262, 263, 264 Likewise, insulin modulates the expression of adiponectin, another well-known adipokine, the abundance of its receptor in adipose and muscle tissue265, 266 as well as its secretion.267, 268 Adiponectin is not only involved in glucose and fatty acid metabolism269 but it also forestalls β-cell apoptosis and induces insulin gene expression and release;270 the latter was mediated by the ERK/Akt pathway in one study270 and by the AMPK pathway in another study.271 Other adipokines, such as apelin,272, 273 chemerin,274, 275, 276 omentin,277, 278 resistin279 and visfatin,280, 281 were also shown to directly interact with insulin, whereas retinol-binding protein 4, tumor necrosis factor-α and vaspin are related to insulin in an indirect manner.282 In addition to adipokine secretion by adipocytes, myocytes release cytokines, which are referred to as myokines. Fibroblast growth factor-21 is a widely expressed protein with a broad mode of action, including the regulation of carbohydrate and fatty acid metabolism283 and may be considered as a myokine due to its secretion from muscle cells.284 Fibroblast growth factor-21 is regulated by insulin285 through the PI3K/Akt1 signaling pathway.286 Interleukin (IL)-6, which is both an adipokine and myokine,287 was shown to influence the pancreas by controlling the expression of pro-glucagon mRNA as well as glucagon secretion. It also increases α-cell proliferation and islet mass while protecting the pancreas from metabolic stress-induced apoptosis.288 Furthermore, IL-6 increased GLP-1 production from proglucagon in pancreatic α-cells and its secretion from α-cells and intestinal L-cells, eventually resulting in a GLP-1-mediated increase in insulin secretion.289
Modulating insulin secretion as a means of diabetes therapy
Due to the worldwide, still spreading epidemic of T2DM, there is an urgent need for (new) anti-diabetic drugs and therapies that are more effective and have fewer side effects. Currently, the most commonly used drugs can be classified into agents that enhance insulin secretion (secretagogues such as sulfonylureas (SUs) and incretin mimetics), sensitize the target organs of insulin (for example, metformin from the class of biguanides or thiazolidinediones), or reduce glucose absorption from the gastrointestinal tract (inhibitors of gastrointestinal α-glucosidase). Different therapies address different problems and stages of T2DM and may be prescribed in combination to exert synergistic effects.
Sulfonylureas
A-glucosidase inhibitors and sensitizers do not target the pancreas or insulin secretion itself but instead target the upstream (slowed intestinal glucose absorption) or downstream (improved insulin sensitivity) processes. In contrast, insulin secretagogues directly modulate insulin release. The SUs are the first broadly applied oral anti-hyperglycemic drugs. To date, there are two generations of agents: acetohexamide, chlorpropamide, tolazamide and tolbutamide, which constitute the first generation and glibenclamide/glyburide, gliclazide, glimepiride, glipizide and gliquidone, which comprise the second generation. First-generation SUs are rarely used these days since tolbutamide intake was associated with an increase in lethal cardiac events.290, 291 More importantly, the second-generation SUs are more potent due to modifications in their side chains’ structure, resulting in improved SUR-affinity, accompanied by lower effective plasma levels, which in turn may reduce undesirable drug-protein interactions.
All SUs share a central SU backbone but differ in their side chains. Despite having different pharmacokinetics, they work in the same way, namely by triggering endogenous insulin release by blocking KATP-channels and hence activating the insulin signaling pathway. More precisely, SUs bind to the sulfonylurea receptor (SUR) subunit of the KATP-channel with high affinity.292, 293 SUR, together with the pore-forming subunit Kir6.x, forms a hetero–octameric complex consisting of four inner Kir6.x subunits surrounded by four SUR subunits (4:4 stoichiometry).294, 295 Moreover, different isoforms of the two subunits are expressed, depending on the tissue-specific expression of the KATP-channels: SUR1 and Kir6.2 are expressed in the pancreas and brain,296 Kir6.2 and SUR2A are expressed in the heart and skeletal muscle,297 while SUR2B is expressed in the brain and smooth muscle,298 and Kir6.1 and SUR2B are expressed in vascular smooth muscle.299 Although SUs bind to both, SURs and Kir6.2, the interactions with the latter are of low affinity300, 301 and hence only SUR-interacting agents are used for diabetes treatment. In addition to their mode of action as inhibitors of KATP-channels, SUs were shown to improve glucose uptake into insulin-dependent tissues and glucose disposal as well as to reduce hepatic glycogenolysis/gluconeogenesis.302, 303, 304
In contrast to SUs inactivating the KATP-channels by binding to the SUR1 subunit, ATP closes them by interacting with Kir6.2.305 Moreover, while the binding of only one ATP molecule is sufficient to completely close the channel,306 inhibition by SUs is incomplete as the channel might still open even when SUs are bound to SUR1.299 Nonetheless, second-generation SUs reduce the glycated hemoglobin or HbA1c, which represent the average plasma glucose concentrations over time and thus serve as a diagnostic measure for diabetes mellitus, by 1.0–2.0%. In addition to the weight gain attributed to the anabolic effects of increased insulin secretion, the main side effect of SUs is hypoglycemia307, 308 due to excess circulating insulin levels and due to the fact that SUs evoke insulin secretion in a glucose-independent manner.309
Although they are not SUs per se, meglitinides, that is, repaglinide and nateglinide, share their mode of action of inhibiting KATP-channels.310 However, meglitinides and some of the second-generation SUs, for example, glibenclamide, interact with both, the SUR1 and the SUR2A or B isoforms.311 Despite the possible disadvantage of this generalized binding that may cause undesirable effects on other KATP-channel types, for example, those in the heart,312 meglitinides, namely nateglinide, have an earlier onset of action and a faster dissociation rate from the sulfonylurea receptor,313, 314, 315 resulting in a diminished risk of hypoglycemia.316 Like SUs, meglitinides also cause weight gain.317, 318
Incretin mimetics
Another group of insulin secretagogues is comprised of the incretins GLP-1 and GIP. As both incretins are rapidly inactivated by the enzyme dipeptidyl peptidase IV (DPP-IV),319 their application in T2DM treatment focuses on modified analogues320, 321, 322, 323, 324, 325 or receptor agonists, including the well-known, short-acting exenatide.326, 327, 328 The long-lasting agonists exenatide LAR,329, 330 liraglutide331, 332 and lixisenatide333, 334, 335 are currently under investigation. However, based on the lipogenetic properties205, 206, 207 of GIP, insufficient insulin-potentiating effects in T2DM patients220, 336 and a possible worsening effect by GIP,337, 338 the focus is on GLP-1 analogues/receptor agonists for T2DM treatment. By acting on its receptor, GLP-1 induces the signaling cascade described under ‘External factors affecting pancreatic hormone secretion’, resulting in its main effect: potentiating insulin secretion. In addition to reducing the HbA1C levels, GLP-1 analogues/receptor agonists promote weight loss and, more importantly, do not evoke hypoglycemia, as do SUs,326, 327, 328, 329, 330, 331, 332, 333, 334 due to the glucose-dependent mode of action and the self-regulating mechanism of GLP-1.68, 336, 339 When blood glucose levels are lowered to physiological levels, GLP-1 is incapable of enhancing insulin secretion, thereby preventing hypoglycemia.79, 340 In addition, GLP-1 (analogues/receptor agonists) exerts further pancreatic and extrapancreatic actions, as mentioned under ‘Interplay between the pancreatic islets and other organs’. Although GLP-1 (analogues/receptor agonists) exhibits some minor side effects, including nausea, vomiting or gastrointestinal impairments,326, 327, 328, 329, 330, 331, 332, 333, 334, 335 the beneficial properties outweigh the negative effects, and thus, GLP-1 is a promising anti-diabetic agent.
Insulin sensitizers
Metformin, which is generally the most widely used first-line anti-diabetic medication,341 is a so-called (insulin) sensitizer. It not only diminishes hepatic glucose output due to glycogenolysis/gluconeogenesis342 but it also enhances glucose uptake into peripheral tissues, such as skeletal muscle, by activating 5′-adenosine monophosphate-activated protein kinase (AMPK-α2).343 Furthermore, it supports weight loss344 by reducing food consumption.345 With respect to its effects on β-cell function, metformin was shown to increase insulin gene expression,346 possibly by nuclear accumulation of pdx1 and its subsequently improved DNA-binding activity.347 Interestingly, metformin exerts opposing effects on β-cell proliferation and/or apoptosis; on the one hand, it suppresses β-cell proliferation and enhances apoptosis through an AMPK-dependent and autophagy-mediated mechanism348 following the metformin-induced activation of c-Jun-N-terminal kinase and caspase-3.349 On the other hand, metformin reduces caspase-3- and -8-mediated apoptosis in isolated islets from T2DM patients350 and protects against lipotoxicity-induced β-cell defects.348, 351
The other members of the sensitizer group include the thiazolidinediones (or glitazones). Currently, only pioglitazone is available; troglitazone was withdrawn from the market in 2000 and rosiglitazone was withdrawn in 2010 due to liver toxicity, drug-induced hepatitis352, 353, 354 and the increased risk of cardiovascular events, respectively.355 Their mode of action involves activation of the peroxisome proliferator-activated receptor (PPARγ), a nuclear transcription factor that is highly expressed in adipose tissue, and the subsequent regulation of genes that are involved in glucose and fat metabolism.356, 357, 358 By promoting lipogenesis, FFAs are removed from the blood stream, whereupon cells become dependent on glucose as an energy substrate. However, enhanced lipogenesis also leads to the weight gain observed in thiazolidinedione-treated T2DM patients.359 In contrast to metformin, pioglitazone prevents (oxidative stress-induced) apoptosis360, 361 by decreasing the expression of apoptosis-promoting genes, while increasing anti-apoptotic and anti-oxidative gene expression. However, this may depend on the disease state.362, 363 Furthermore, pioglitazone increases β-cell mass by upregulating cell differentiation/proliferation genes.364 Although they have partially different modes of action, both groups of sensitizers cause a reduction in the HbA1c level by 1.5–2.0%.
A-glucosidase inhibitors
A-glucosidase inhibitors, such as acarbose, miglitol and voglibose, not only decelerate the breakdown of starch into glucose in the small intestine but also decrease its bioavailability, resulting in reduced levels of glucose entering the blood stream and hence attenuated postprandial glucose excursions.365, 366, 367, 368, 369, 370 In addition, they support weight loss371, 372 and ameliorate blood pressure,373 insulin sensitivity367, 368 and triglyceride levels.369, 370 Similar to pioglitazone, α-glucosidase inhibitors attenuate reductions in β-cell mass, which may delay the onset of diabetes.374, 375, 376 As α-glucosidase inhibitors only mildly reduce HbA1c levels (0.5–1.0%), they are usually only used in the early stage of T2DM, that is, impaired glucose tolerance or in combination with other drugs.377
Conclusions and outlook
The pancreas has key roles in maintaining normal blood glucose levels by producing and releasing insulin and glucagon. These opponents interact not only with each other through the intra-islet insulin axis378, 379, 380, 381 but also with other organs/tissues, that is, the brain, liver, gut as well as insulin-dependent adipose and muscle tissues. Altogether, the islet–organ/tissues axes described here form a highly sophisticated network that includes, but is not limited to, various signaling molecules, that is, neuropeptides (brain-derived neurotrophic factor, NPY, melanin concentrating hormone, gastrin-releasing peptide, VIP and PACAP), hepatokines (betatrophin and HNFs), enteroendocrine hormones (the incretins GLP-1 and GIP, the decretins NmU and limostatin, gastrin and CCK) as well as adipokines (leptin and adiponectin) and myokines (fibroblast growth factor-21 and IL-6) that mainly interact through GPCR signaling pathways, such as the cAMP cascade. In good health, the well-functioning interactions between all of the organs and tissues involved ensure glucose homeostasis. However, impairments in the secretion of and/or sensitivity to insulin may result in metabolic diseases, such as T2DM. Referring to the American Diabetes Association, T2DM and noninsulin-dependent diabetes mellitus are characterized by insulin resistance, hyperglycemia and a relative insulin deficiency. Furthermore, T2DM is associated with low-grade inflammation,382, 383 cardiovascular disease,384, 385 nephropathy386, 387 and alterations in the secretion of various hormones, including IL-6, IL-18, tumor necrosis factor-α,388 adiponectin and leptin,389 neuropeptides,390 ghrelin391, 392 and the incretins GLP-1 and GIP.219, 220, 221, 222, 223 Although lifestyle interventions393 and weight loss394 reverse T2DM in early stages, when insulin is still secreted, T2DM patients may become dependent on anti-diabetic drugs in later stages. Currently, there are three classes of agents: insulin secretatogues, insulin sensitizers and α-glucosidase inhibitors, all of which have different modes of action and hence target different stages and symptoms of T2DM. Treatments that modulate insulin release—on condition of an appropriate insulin sensitivity of the target organs—appear to be promising approaches. Current research is unveiling new molecules, enzymes and interactions that are involved in the signaling pathways underlying insulin secretion, among others, and is likely to introduce new therapeutic approaches. Strategies that target these mediating molecules may include, but are not limited to, the calcium sensor Syt-7,90, 395 the SNARE-associated protein Snapin,89 the t-SNARE SNAP-25,396 cyclin-dependent kinase (Cdk) 5,397 ryanodine receptor (RyR) 2,398 the nucleotide exchange factor and intracellular cAMP sensor Epac2,57, 58, 59, 399 mammalian uncoordinated proteins (munc)13400, 401 and munc1840, 41 as well as the Ras-related proteins (Rab) 3A402 and 27A.403
References
Chandra R, Liddle RA . Neural and hormonal regulation of pancreatic secretion. Curr Opin Gastroenterol 2009; 25: 441–446.
Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005; 53: 1087–1097.
Katsuura G, Asakawa A, Inui A . Roles of pancreatic polypeptide in regulation of food intake. Peptides 2002; 23: 323–329.
Wierup N, Svensson H, Mulder H, Sundler F . The ghrelin cell: a novel developmentally regulated islet cell in the human pancreas. Regul Pept 2002; 107: 63–69.
Goke B . Islet cell function: alpha and beta cells—partners towards normoglycaemia. Int J Clin Pract Suppl 2008; 159: 2–7.
Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ et al. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 2009; 58: 403–411.
Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M et al. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab 2003; 88: 3989–3992.
Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A . The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA 2006; 103: 2334–2339.
Freychet L, Rizkalla SW, Desplanque N, Basdevant A, Zirinis P, Tchobroutsky G et al. Effect of intranasal glucagon on blood glucose levels in healthy subjects and hypoglycaemic patients with insulin-dependent diabetes. Lancet 1988; 1: 1364–1366.
Komatsu M, Takei M, Ishii H, Sato Y . Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig 2013; 4: 511–516.
Khan AH, Pessin JE . Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia 2002; 45: 1475–1483.
Kohn AD, Summers SA, Birnbaum MJ, Roth RA . Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 1996; 271: 31372–31378.
Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med 2000; 6: 924–928.
Sibrowski W, Seitz HJ . Rapid action of insulin and cyclic AMP in the regulation of functional messenger RNA coding for glucokinase in rat liver. J Biol Chem 1984; 259: 343–346.
Kim SY, Kim HI, Kim TH, Im SS, Park SK, Lee IK et al. SREBP-1c mediates the insulin-dependent hepatic glucokinase expression. J Biol Chem 2004; 279: 30823–30829.
Aiston S, Hampson LJ, Arden C, Iynedjian PB, Agius L . The role of protein kinase B/Akt in insulin-induced inactivation of phosphorylase in rat hepatocytes. Diabetologia 2006; 49: 174–182.
Syed NA, Khandelwal RL . Reciprocal regulation of glycogen phosphorylase and glycogen synthase by insulin involving phosphatidylinositol-3 kinase and protein phosphatase-1 in HepG2 cells. Mol Cell Biochem 2000; 211: 123–136.
Miller TB Jr, Larner J . Mechanism of control of hepatic glycogenesis by insulin. J Biol Chem 1973; 248: 3483–3488.
Akpan JO, Gardner R, Wagle SR . Studies on the effects of insulin and acetylcholine on activation of glycogen synthase and on glycogenesis in hepatocytes. Biochem Biophys Res Commun 1974; 61: 222–229.
Stalmans W, De Wulf H, Hue L, Hers HG . The sequential inactivation of glycogen phosphorylase and activation of glycogen synthetase in liver after the administration of glucose to mice and rats. The mechanism of the hepatic threshold to glucose. Eur J Biochem 1974; 41: 127–134.
O'Brien RM, Lucas PC, Forest CD, Magnuson MA, Granner DK . Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science 1990; 249: 533–537.
Streeper RS, Svitek CA, Chapman S, Greenbaum LE, Taub R, O'Brien RM . A multicomponent insulin response sequence mediates a strong repression of mouse glucose-6-phosphatase gene transcription by insulin. J Biol Chem 1997; 272: 11698–11701.
Duong DT, Waltner-Law ME, Sears R, Sealy L, Granner DK . Insulin inhibits hepatocellular glucose production by utilizing liver-enriched transcriptional inhibitory protein to disrupt the association of CREB-binding protein and RNA polymerase II with the phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 2002; 277: 32234–32242.
Nakae J, Kitamura T, Silver DL, Accili D . The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 2001; 108: 1359–1367.
Schmoll D, Walker KS, Alessi DR, Grempler R, Burchell A, Guo S et al. Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J Biol Chem 2000; 275: 36324–36333.
Lochhead PA, Coghlan M, Rice SQ, Sutherland C . Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes 2001; 50: 937–946.
Walton PE, Etherton TD . Stimulation of lipogenesis by insulin in swine adipose tissue: antagonism by porcine growth hormone. J Anim Sci 1986; 62: 1584–1595.
McTernan PG, Harte AL, Anderson LA, Green A, Smith SA, Holder JC et al. Insulin and rosiglitazone regulation of lipolysis and lipogenesis in human adipose tissue in vitro. Diabetes 2002; 51: 1493–1498.
Biolo G, Declan Fleming RY, Wolfe RR . Physiologic hyperinsulinemia stimulates protein synthesis and enhances transport of selected amino acids in human skeletal muscle. J Clin Invest 1995; 95: 811–819.
Ashcroft FM, Proks P, Smith PA, Ammala C, Bokvist K, Rorsman P . Stimulus-secretion coupling in pancreatic beta cells. J Cell Biochem 1994; 55 Suppl: 54–65.
Henquin JC . Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000; 49: 1751–1760.
Cerasi E, Luft R . The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol (Copenh) 1967; 55: 278–304.
Porte D Jr, Pupo AA . Insulin responses to glucose: evidence for a two pool system in man. J Clin Invest 1969; 48: 2309–2319.
Curry DL, Bennett LL, Grodsky GM . Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968; 83: 572–584.
Hao M, Li X, Rizzo MA, Rocheleau JV, Dawant BM, Piston DW . Regulation of two insulin granule populations within the reserve pool by distinct calcium sources. J Cell Sci 2005; 118 (Pt 24): 5873–5884.
Olofsson CS, Gopel SO, Barg S, Galvanovskis J, Ma X, Salehi A et al. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch 2002; 444: 43–51.
Thurmond DC . Regulation of Insulin Action and Insulin Secretion by SNARE-Mediated Vesicle Exocytosis. Landes Bioscience: Austin, TX, USA, 2000.
Chapman ER, An S, Barton N, Jahn R . SNAP-25, a t-SNARE which binds to both syntaxin and synaptobrevin via domains that may form coiled coils. J Biol Chem 1994; 269: 27427–27432.
Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT . Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J Biol Chem 1997; 272: 28036–28041.
Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J et al. Munc18-1 promotes large dense-core vesicle docking. Neuron 2001; 31: 581–591.
Lam PP, Ohno M, Dolai S, He Y, Qin T, Liang T et al. Munc18b is a major mediator of insulin exocytosis in rat pancreatic beta-cells. Diabetes 2013; 62: 2416–2428.
Foster LJ, Yeung B, Mohtashami M, Ross K, Trimble WS, Klip A . Binary interactions of the SNARE proteins syntaxin-4, SNAP23, and VAMP-2 and their regulation by phosphorylation. Biochemistry 1998; 37: 11089–11096.
Ravichandran V, Chawla A, Roche PA . Identification of a novel syntaxin- and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J Biol Chem 1996; 271: 13300–13303.
Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY . SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins. Endocr Rev 2007; 28: 653–663.
Regazzi R, Wollheim CB, Lang J, Theler JM, Rossetto O, Montecucco C et al. VAMP-2 and cellubrevin are expressed in pancreatic beta-cells and are essential for Ca(2+)-but not for GTP gamma S-induced insulin secretion. EMBO J 1995; 14: 2723–2730.
Zhu D, Koo E, Kwan E, Kang Y, Park S, Xie H et al. Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia 2013; 56: 359–369.
Zhu D, Zhang Y, Lam PP, Dolai S, Liu Y, Cai EP et al. Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth. Cell Metab 2012; 16: 238–249.
Gustavsson N, Han W . Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci Rep 2009; 29: 245–259.
Gut A, Kiraly CE, Fukuda M, Mikoshiba K, Wollheim CB, Lang J . Expression and localisation of synaptotagmin isoforms in endocrine beta-cells: their function in insulin exocytosis. J Cell Sci 2001; 114 (Pt 9): 1709–1716.
Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK et al. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+ -induced glucagon exocytosis in pancreas. J Physiol 2009; 587 (Pt 6): 1169–1178.
Mizuta M, Kurose T, Miki T, Shoji-Kasai Y, Takahashi M, Seino S et al. Localization and functional role of synaptotagmin III in insulin secretory vesicles in pancreatic beta-cells. Diabetes 1997; 46: 2002–2006.
Iezzi M, Kouri G, Fukuda M, Wollheim CB . Synaptotagmin V and IX isoforms control Ca2+ -dependent insulin exocytosis. J Cell Sci 2004; 117 (Pt 15): 3119–3127.
Sadana R, Dessauer CW . Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals 2009; 17: 5–22.
Das R, Esposito V, Abu-Abed M, Anand GS, Taylor SS, Melacini G . cAMP activation of PKA defines an ancient signaling mechanism. Proc Natl Acad Sci USA 2007; 104: 93–98.
Li S, Tsalkova T, White MA, Mei FC, Liu T, Wang D et al. Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J Biol Chem 2011; 286: 17889–17897.
Leech CA, Chepurny OG, Holz GG . Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam Horm 2010; 84: 279–302.
Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL et al. Epac-selective cAMP analog 8-pCPT-2'-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem 2003; 278: 8279–8285.
Dzhura I, Chepurny OG, Kelley GG, Leech CA, Roe MW, Dzhura E et al. Epac2-dependent mobilization of intracellular Ca(2)+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in beta-cells of phospholipase C-epsilon knockout mice. J Physiol 2010; 588 (Pt 24): 4871–4889.
Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333–19338.
Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S . PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J 1999; 18: 4722–4732.
Ribalet B, Ciani S, Eddlestone GT . ATP mediates both activation and inhibition of K(ATP) channel activity via cAMP-dependent protein kinase in insulin-secreting cell lines. J Gen Physiol 1989; 94: 693–717.
Kanno T, Suga S, Wu J, Kimura M, Wakui M . Intracellular cAMP potentiates voltage-dependent activation of L-type Ca2+ channels in rat islet beta-cells. Pflugers Arch 1998; 435: 578–580.
Safayhi H, Haase H, Kramer U, Bihlmayer A, Roenfeldt M, Ammon HP et al. L-type calcium channels in insulin-secreting cells: biochemical characterization and phosphorylation in RINm5F cells. Mol Endocrinol 1997; 11: 619–629.
Wan QF, Dong Y, Yang H, Lou X, Ding J, Xu T . Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+. J Gen Physiol 2004; 124: 653–662.
Renstrom E, Eliasson L, Rorsman P . Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol 1997; 502 (Pt 1): 105–118.
Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM . Glucose sensing in L cells: a primary cell study. Cell Metab 2008; 8: 532–539.
Parker HE, Habib AM, Rogers GJ, Gribble FM, Reimann F . Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009; 52: 289–298.
Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V . Glucagon-like peptide-1 (7-36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrinol 1993; 138: 159–166.
Kuhre RE, Gribble FM, Hartmann B, Reimann F, Windelov JA, Rehfeld JF et al. Fructose stimulates GLP-1 but not GIP secretion in mice, rats, and humans. Am J Physiol Gastrointest Liver Physiol 2014; 306: G622–G630.
Reimann F, Williams L, da Silva Xavier G, Rutter GA, Gribble FM . Glutamine potently stimulates glucagon-like peptide-1 secretion from GLUTag cells. Diabetologia 2004; 47: 1592–1601.
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012; 61: 364–371.
Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med 2005; 11: 90–94.
Schauder P, Brown JC, Frerichs H, Creutzfeldt W . Gastric inhibitory polypeptide: effect on glucose-induced insulin release from isolated rat pancreatic islets in vitro. Diabetologia 1975; 11: 483–484.
Dupre J, Ross SA, Watson D, Brown JC . Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 1973; 37: 826–828.
Kreymann B, Williams G, Ghatei MA, Bloom SR . Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987; 2: 1300–1304.
Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, Ling Z et al. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 1996; 45: 257–261.
Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF . Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA 1987; 84: 3434–3438.
Ramos LS, Zippin JH, Kamenetsky M, Buck J, Levin LR . Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J Gen Physiol 2008; 132: 329–338.
Holz GGt 4th, Leech CA, Habener JF . Activation of a cAMP-regulated Ca(2+)-signaling pathway in pancreatic beta-cells by the insulinotropic hormone glucagon-like peptide-1. J Biol Chem 1995; 270: 17749–17757.
Wheeler MB, Gelling RW, McIntosh CH, Georgiou J, Brown JC, Pederson RA . Functional expression of the rat pancreatic islet glucose-dependent insulinotropic polypeptide receptor: ligand binding and intracellular signaling properties. Endocrinology 1995; 136: 4629–4639.
Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE 3rd . Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology 1993; 133: 57–62.
Volz A, Goke R, Lankat-Buttgereit B, Fehmann HC, Bode HP, Goke B . Molecular cloning, functional expression, and signal transduction of the GIP-receptor cloned from a human insulinoma. FEBS Lett 1995; 373: 23–29.
Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF . cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic beta-cells. A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7-37). J Biol Chem 1999; 274: 14147–14156.
Gromada J, Dissing S, Bokvist K, Renstrom E, Frokjaer-Jensen J, Wulff BS et al. Glucagon-like peptide I increases cytoplasmic calcium in insulin-secreting beta TC3-cells by enhancement of intracellular calcium mobilization. Diabetes 1995; 44: 767–774.
Lu M, Wheeler MB, Leng XH, Boyd AE 3rd . The role of the free cytosolic calcium level in beta-cell signal transduction by gastric inhibitory polypeptide and glucagon-like peptide I(7-37). Endocrinology 1993; 132: 94–100.
Fridolf T, Ahren B . Effects of glucagon like peptide-1(7-36) amide on the cytoplasmic Ca(2+)-concentration in rat islet cells. Mol Cell Endocrinol 1993; 96: 85–90.
Cullinan CA, Brady EJ, Saperstein R, Leibowitz MD . Glucose-dependent alterations of intracellular free calcium by glucagon-like peptide-1(7-36amide) in individual ob/ob mouse beta-cells. Cell Calcium 1994; 15: 391–400.
Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C . Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes 1993; 42: 1678–1682.
Song WJ, Seshadri M, Ashraf U, Mdluli T, Mondal P, Keil M et al. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metab 2011; 13: 308–319.
Wu B, Wei S, Petersen N, Ali Y, Wang X, Bacaj T et al. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proc Natl Acad Sci USA 2015; 112: 9996–10001.
Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD . Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun 2005; 335: 97–104.
Fujiwara K, Maekawa F, Yada T . Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: mediation by PLC and L-type Ca2+ channel and link to insulin release. Am J Physiol Endocrinol Metab 2005; 289: E670–E677.
Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C et al. Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res 2005; 322: 207–215.
Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003; 422: 173–176.
Ximenes HM, Hirata AE, Rocha MS, Curi R, Carpinelli AR . Propionate inhibits glucose-induced insulin secretion in isolated rat pancreatic islets. Cell Biochem Funct 2007; 25: 173–178.
Porte D Jr, Williams RH . Inhibition of insulin release by norepinephrine in man. Science 1966; 152: 1248–1250.
Peterhoff M, Sieg A, Brede M, Chao CM, Hein L, Ullrich S . Inhibition of insulin secretion via distinct signaling pathways in alpha2-adrenoceptor knockout mice. Eur J Endocrinol 2003; 149: 343–350.
Porte D Jr . A receptor mechanism for the inhibition of insulin release by epinephrine in man. J Clin Invest 1967; 46: 86–94.
Rossi J, Santamaki P, Airaksinen MS, Herzig KH . Parasympathetic innervation and function of endocrine pancreas requires the glial cell line-derived factor family receptor alpha2 (GFRalpha2). Diabetes 2005; 54: 1324–1330.
Yoshimatsu H, Niijima A, Oomura Y, Yamabe K, Katafuchi T . Effects of hypothalamic lesion on pancreatic autonomic nerve activity in the rat. Brain Res 1984; 303: 147–152.
Borden P, Houtz J, Leach SD, Kuruvilla R . Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep 2013; 4: 287–301.
Hopkins DF, Williams G . Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet Med 1997; 14: 1044–1050.
Ho L, Yemul S, Knable L, Katsel P, Zhao R, Haroutunian V et al. Insulin receptor expression and activity in the brains of nondiabetic sporadic Alzheimer's disease cases. Int J Alzheimers Dis 2012; 2012: 321280.
Hill JM, Lesniak MA, Pert CB, Roth J . Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience 1986; 17: 1127–1138.
Marks JL, Porte D Jr, Stahl WL, Baskin DG . Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990; 127: 3234–3236.
Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L . Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci 2002; 5: 566–572.
Young WS 3rd . Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides 1986; 8: 93–97.
Mirshamsi S, Laidlaw HA, Ning K, Anderson E, Burgess LA, Gray A et al. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci 2004; 5: 54.
Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004; 53: 1959–1965.
Berthoud HR, Jeanrenaud B . Acute hyperinsulinemia and its reversal by vagotomy after lesions of the ventromedial hypothalamus in anesthetized rats. Endocrinology 1979; 105: 146–151.
Rohner-Jeanrenaud F, Jeanrenaud B . Consequences of ventromedial hypothalamic lesions upon insulin and glucagon secretion by subsequently isolated perfused pancreases in the rat. J Clin Invest 1980; 65: 902–910.
Goto Y, Carpenter RG, Berelowitz M, Frohman LA . Effect of ventromedial hypothalamic lesions on the secretion of somatostatin, insulin, and glucagon by the perfused rat pancreas. Metabolism 1980; 29: 986–990.
Rohner F, Dufour AC, Karakash C, Le Marchand Y, Ruf KB, Jeanrenaud B . Immediate effect of lesion of the ventromedial hypothalamic area upon glucose-induced insulin secretion in anaesthetized rats. Diabetologia 1977; 13: 239–242.
Hanyu O, Yamatani K, Ikarashi T, Soda S, Maruyama S, Kamimura T et al. Brain-derived neurotrophic factor modulates glucagon secretion from pancreatic alpha cells: its contribution to glucose metabolism. Diabetes Obes Metab 2003; 5: 27–37.
Gotoh K, Masaki T, Chiba S, Ando H, Fujiwara K, Shimasaki T et al. Hypothalamic brain-derived neurotrophic factor regulates glucagon secretion mediated by pancreatic efferent nerves. J Neuroendocrinol 2013; 25: 302–311.
Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD . The central melanocortin system can directly regulate serum insulin levels. Endocrinology 2000; 141: 3072–3079.
Malaisse W, Malaisse-Lagae F, Wright PH, Ashmore J . Effects of adrenergic and cholinergic agents upon insulin secretion in vitro. Endocrinology 1967; 80: 975–978.
Vettor R, Pagano C, Granzotto M, Englaro P, Angeli P, Blum WF et al. Effects of intravenous neuropeptide Y on insulin secretion and insulin sensitivity in skeletal muscle in normal rats. Diabetologia 1998; 41: 1361–1367.
Moltz JH, McDonald JK . Neuropeptide Y: direct and indirect action on insulin secretion in the rat. Peptides 1985; 6: 1155–1159.
Imai Y, Patel HR, Hawkins EJ, Doliba NM, Matschinsky FM, Ahima RS . Insulin secretion is increased in pancreatic islets of neuropeptide Y-deficient mice. Endocrinology 2007; 148: 5716–5723.
Morgan DG, Kulkarni RN, Hurley JD, Wang ZL, Wang RM, Ghatei MA et al. Inhibition of glucose stimulated insulin secretion by neuropeptide Y is mediated via the Y1 receptor and inhibition of adenylyl cyclase in RIN 5AH rat insulinoma cells. Diabetologia 1998; 41: 1482–1491.
Schwetz TA, Ustione A, Piston DW . Neuropeptide Y and somatostatin inhibit insulin secretion through different mechanisms. Am J Physiol Endocrinol Metab 2013; 304: E211–E221.
Gautam D, Han SJ, Hamdan FF, Jeon J, Li B, Li JH et al. A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab 2006; 3: 449–461.
Schebalin M, Said SI, Makhlouf GM . Stimulation of insulin and glucagon secretion by vasoactive intestinal peptide. Am J Physiol 1977; 232: E197–E200.
Filipsson K, Tornoe K, Holst J, Ahren B . Pituitary adenylate cyclase-activating polypeptide stimulates insulin and glucagon secretion in humans. J Clin Endocrinol Metab 1997; 82: 3093–3098.
Pissios P, Ozcan U, Kokkotou E, Okada T, Liew CW, Liu S et al. Melanin concentrating hormone is a novel regulator of islet function and growth. Diabetes 2007; 56: 311–319.
Borboni P, Porzio O, Pierucci D, Cicconi S, Magnaterra R, Federici M et al. Molecular and functional characterization of pituitary adenylate cyclase-activating polypeptide (PACAP-38)/vasoactive intestinal polypeptide receptors in pancreatic beta-cells and effects of PACAP-38 on components of the insulin secretory system. Endocrinology 1999; 140: 5530–5537.
Straub SG, Sharp GW . A wortmannin-sensitive signal transduction pathway is involved in the stimulation of insulin release by vasoactive intestinal polypeptide and pituitary adenylate cyclase-activating polypeptide. J Biol Chem 1996; 271: 1660–1668.
Gregersen S, Ahren B . Studies on the mechanisms by which gastrin releasing peptide potentiates glucose-induced insulin secretion from mouse islets. Pancreas 1996; 12: 48–57.
Pettersson M, Ahren B . Gastrin releasing peptide (GRP): effects on basal and stimulated insulin and glucagon secretion in the mouse. Peptides 1987; 8: 55–60.
Bellisle F, Louis-Sylvestre J, Demozay F, Blazy D, Le Magnen J . Cephalic phase of insulin secretion and food stimulation in humans: a new perspective. Am J Physiol 1985; 249 (6 Pt 1): E639–E645.
Woods SC, Alexander KR, Porte D Jr . Conditioned insulin secretion and hypoglycemia following repeated injections of tolbutamide in rats. Endocrinology 1972; 90: 227–231.
Stockhorst U, Steingruber HJ, Scherbaum WA . Classically conditioned responses following repeated insulin and glucose administration in humans. Behav Brain Res 2000; 110: 143–159.
Berthoud HR, Jeanrenaud B . Sham feeding-induced cephalic phase insulin release in the rat. Am J Physiol 1982; 242: E280–E285.
Power ML, Schulkin J . Anticipatory physiological regulation in feeding biology: cephalic phase responses. Appetite 2008; 50: 194–206.
Ahren B, Holst JJ . The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes 2001; 50: 1030–1038.
Powley TL . Vagal circuitry mediating cephalic-phase responses to food. Appetite 2000; 34: 184–188.
Banks WA, Jaspan JB, Kastin AJ . Selective, physiological transport of insulin across the blood-brain barrier: novel demonstration by species-specific radioimmunoassays. Peptides 1997; 18: 1257–1262.
Woods SC, Lotter EC, McKay LD, Porte D Jr . Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979; 282: 503–505.
Woods SC, Stein LJ, McKay LD, Porte D Jr . Suppression of food intake by intravenous nutrients and insulin in the baboon. Am J Physiol 1984; 247 (2 Pt 2): R393–R401.
Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ et al. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci 2002; 22: 9048–9052.
Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 2003; 52: 227–231.
Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS . PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 2005; 115: 951–958.
Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A et al. Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology 1992; 130: 3608–3616.
Flood JF, Morley JE . Increased food intake by neuropeptide Y is due to an increased motivation to eat. Peptides 1991; 12: 1329–1332.
Morley JE, Hernandez EN, Flood JF . Neuropeptide Y increases food intake in mice. Am J Physiol 1987; 253 (3 Pt 2): R516–R522.
Dube MG, Xu B, Crowley WR, Kalra PS, Kalra SP . Evidence that neuropeptide Y is a physiological signal for normal food intake. Brain Res 1994; 646: 341–344.
Tang-Christensen M, Vrang N, Ortmann S, Bidlingmaier M, Horvath TL, Tschop M . Central administration of ghrelin and agouti-related protein (83-132) increases food intake and decreases spontaneous locomotor activity in rats. Endocrinology 2004; 145: 4645–4652.
Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D et al. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 1998; 139: 4428–4431.
Schwartz MW, Marks JL, Sipols AJ, Baskin DG, Woods SC, Kahn SE et al. Central insulin administration reduces neuropeptide Y mRNA expression in the arcuate nucleus of food-deprived lean (Fa/Fa) but not obese (fa/fa) Zucker rats. Endocrinology 1991; 128: 2645–2647.
Carvalheira JB, Ribeiro EB, Araujo EP, Guimaraes RB, Telles MM, Torsoni M et al. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia 2003; 46: 1629–1640.
Ikeda H, West DB, Pustek JJ, Figlewicz DP, Greenwood MR, Porte D Jr et al. Intraventricular insulin reduces food intake and body weight of lean but not obese Zucker rats. Appetite 1986; 7: 381–386.
Obici S, Zhang BB, Karkanias G, Rossetti L . Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002; 8: 1376–1382.
Gelling RW, Morton GJ, Morrison CD, Niswender KD, Myers MG Jr, Rhodes CJ et al. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab 2006; 3: 67–73.
Jiang G, Zhang BB . Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 2003; 284: E671–E678.
Hers HG . Mechanisms of blood glucose homeostasis. J Inherit Metab Dis 1990; 13: 395–410.
Ramachandran C, Goris J, Waelkens E, Merlevede W, Walsh DA . The interrelationship between cAMP-dependent alpha and beta subunit phosphorylation in the regulation of phosphorylase kinase activity. Studies using subunit specific phosphatases. J Biol Chem 1987; 262: 3210–3218.
Rath VL, Ammirati M, LeMotte PK, Fennell KF, Mansour MN, Danley DE et al. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol Cell 2000; 6: 139–148.
Gao H, Leary JA . Kinetic measurements of phosphoglucomutase by direct analysis of glucose-1-phosphate and glucose-6-phosphate using ion/molecule reactions and Fourier transform ion cyclotron resonance mass spectrometry. Anal Biochem 2004; 329: 269–275.
Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001; 413: 179–183.
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001; 413: 131–138.
Pilkis SJ, El-Maghrabi MR, McGrane M, Pilkis J, Claus TH . Regulation by glucagon of hepatic pyruvate kinase, 6-phosphofructo 1-kinase, and fructose-1,6-bisphosphatase. Fed Proc 1982; 41: 2623–2628.
Rosa JL, Ventura F, Tauler A, Bartrons R . Regulation of hepatic 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase gene expression by glucagon. J Biol Chem 1993; 268: 22540–22545.
Blair JB, Cimbala MA, Foster JL, Morgan RA . Hepatic pyruvate kinase. Regulation by glucagon, cyclic adenosine 3'-5'-monophosphate, and insulin in the perfused rat liver. J Biol Chem 1976; 251: 3756–3762.
Feliu JE, Hue L, Hers HG . Hormonal control of pyruvate kinase activity and of gluconeogenesis in isolated hepatocytes. Proc Natl Acad Sci USA 1976; 73: 2762–2766.
Riou JP, Claus TH, Pilkis SJ . Control of pyruvate kinase activity by glucagon in isolated hepatocytes. Biochem Biophys Res Commun 1976; 73: 591–599.
Decaux JF, Antoine B, Kahn A . Regulation of the expression of the L-type pyruvate kinase gene in adult rat hepatocytes in primary culture. J Biol Chem 1989; 264: 11584–11590.
Vaulont S, Munnich A, Decaux JF, Kahn A . Transcriptional and post-transcriptional regulation of L-type pyruvate kinase gene expression in rat liver. J Biol Chem 1986; 261: 7621–7625.
de Wulf H, Hers HG . The role of glucose, glucagon and glucocorticoids in the regulation of liver glycogen synthesis. Eur J Biochem 1968; 6: 558–564.
Hutson NJ, Brumley FT, Assimacopoulos FD, Harper SC, Exton JH . Studies on the alpha-adrenergic activation of hepatic glucose output. I. Studies on the alpha-adrenergic activation of phosphorylase and gluconeogenesis and inactivation of glycogen synthase in isolated rat liver parenchymal cells. J Biol Chem 1976; 251: 5200–5208.
Ciudad C, Camici M, Ahmad Z, Wang Y, DePaoli-Roach AA, Roach PJ . Control of glycogen synthase phosphorylation in isolated rat hepatocytes by epinephrine, vasopressin and glucagon. Eur J Biochem 1984; 142: 511–520.
Aiston S, Coghlan MP, Agius L . Inactivation of phosphorylase is a major component of the mechanism by which insulin stimulates hepatic glycogen synthesis. Eur J Biochem 2003; 270: 2773–2781.
Hale MA, Kagami H, Shi L, Holland AM, Elsasser HP, Hammer RE et al. The homeodomain protein PDX1 is required at mid-pancreatic development for the formation of the exocrine pancreas. Dev Biol 2005; 286: 225–237.
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H . beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 1998; 12: 1763–1768.
Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M et al. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol 1997; 17: 6002–6013.
Pontoglio M, Sreenan S, Roe M, Pugh W, Ostrega D, Doyen A et al. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J Clin Invest 1998; 101: 2215–2222.
Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki PJ, Boriraj VV et al. Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum Mol Genet 1997; 6: 583–586.
Yi P, Park JS, Melton DA . Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 2013; 153: 747–758.
Wang Y, Quagliarini F, Gusarova V, Gromada J, Valenzuela DM, Cohen JC et al. Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc Natl Acad Sci USA 2013; 110: 16109–16114.
Gusarova V, Alexa CA, Na E, Stevis PE, Xin Y, Bonner-Weir S et al. ANGPTL8/betatrophin does not control pancreatic beta cell expansion. Cell 2014; 159: 691–696.
Jiao Y, Le Lay J, Yu M, Naji A, Kaestner KH . Elevated mouse hepatic betatrophin expression does not increase human beta-cell replication in the transplant setting. Diabetes 2014; 63: 1283–1288.
Hu H, Sun W, Yu S, Hong X, Qian W, Tang B et al. Increased circulating levels of betatrophin in newly diagnosed type 2 diabetic patients. Diabetes Care 2014; 37: 2718–2722.
Chen X, Lu P, He W, Zhang J, Liu L, Yang Y et al. Circulating betatrophin levels are increased in patients with type 2 diabetes and associated with insulin resistance. J Clin Endocrinol Metab 2015; 100: E96–E100.
Fu Z, Berhane F, Fite A, Seyoum B, Abou-Samra AB, Zhang R . Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci Rep 2014; 4: 5013.
Gomez-Ambrosi J, Pascual E, Catalan V, Rodriguez A, Ramirez B, Silva C et al. Circulating betatrophin concentrations are decreased in human obesity and type 2 diabetes. J Clin Endocrinol Metab 2014; 99: E2004–E2009.
Fu Z, Abou-Samra AB, Zhang R . An explanation for recent discrepancies in levels of human circulating betatrophin. Diabetologia 2014; 57: 2232–2234.
Buteau J, Roduit R, Susini S, Prentki M . Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia 1999; 42: 856–864.
Buteau J, Foisy S, Joly E, Prentki M . Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003; 52: 124–132.
Perfetti R, Zhou J, Doyle ME, Egan JM . Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology 2000; 141: 4600–4605.
Trumper A, Trumper K, Trusheim H, Arnold R, Goke B, Horsch D . Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol 2001; 15: 1559–1570.
Ehses JA, Pelech SL, Pederson RA, McIntosh CH . Glucose-dependent insulinotropic polypeptide activates the Raf-Mek1/2-ERK1/2 module via a cyclic AMP/cAMP-dependent protein kinase/Rap1-mediated pathway. J Biol Chem 2002; 277: 37088–37097.
Kim SJ, Winter K, Nian C, Tsuneoka M, Koda Y, McIntosh CH . Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J Biol Chem 2005; 280: 22297–22307.
Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ . Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem 2003; 278: 471–478.
Trumper A, Trumper K, Horsch D . Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol 2002; 174: 233–246.
Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M . Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 2004; 47: 806–815.
Prigeon RL, Quddusi S, Paty B, D'Alessio DA . Suppression of glucose production by GLP-1 independent of islet hormones: a novel extrapancreatic effect. Am J Physiol Endocrinol Metab 2003; 285: E701–E707.
Degn KB, Juhl CB, Sturis J, Jakobsen G, Brock B, Chandramouli V et al. ONE week's treatment with the long-acting glucagon-like peptide 1 derivative liraglutide (NN2211) markedly improves 24-h glycemia and alpha- and beta-cell function and reduces endogenous glucose release in patients with type 2 diabetes. Diabetes 2004; 53: 1187–1194.
Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA . Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab 1996; 81: 327–332.
Zander M, Madsbad S, Madsen JL, Holst JJ . Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 2002; 359: 824–830.
Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol 1999; 276 (5 Pt 2): R1541–R1544.
Kjems LL, Holst JJ, Volund A, Madsbad S . The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 2003; 52: 380–386.
Chang AM, Jakobsen G, Sturis J, Smith MJ, Bloem CJ, An B et al. The GLP-1 derivative NN2211 restores beta-cell sensitivity to glucose in type 2 diabetic patients after a single dose. Diabetes 2003; 52: 1786–1791.
Egan JM, Meneilly GS, Habener JF, Elahi D . Glucagon-like peptide-1 augments insulin-mediated glucose uptake in the obese state. J Clin Endocrinol Metab 2002; 87: 3768–3773.
Meneilly GS, McIntosh CH, Pederson RA, Habener JF, Gingerich R, Egan JM et al. Effect of glucagon-like peptide 1 on non-insulin-mediated glucose uptake in the elderly patient with diabetes. Diabetes Care 2001; 24: 1951–1956.
Eckel RH, Fujimoto WY, Brunzell JD . Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes 1979; 28: 1141–1142.
Song DH, Getty-Kaushik L, Tseng E, Simon J, Corkey BE, Wolfe MM . Glucose-dependent insulinotropic polypeptide enhances adipocyte development and glucose uptake in part through Akt activation. Gastroenterology 2007; 133: 1796–1805.
Kim SJ, Nian C, McIntosh CH . Activation of lipoprotein lipase by glucose-dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP-activated protein kinase cascade. J Biol Chem 2007; 282: 8557–8567.
Bollag RJ, Zhong Q, Ding KH, Phillips P, Zhong L, Qin F et al. Glucose-dependent insulinotropic peptide is an integrative hormone with osteotropic effects. Mol Cell Endocrinol 2001; 177: 35–41.
Xie D, Cheng H, Hamrick M, Zhong Q, Ding KH, Correa D et al. Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone 2005; 37: 759–769.
Xie D, Zhong Q, Ding KH, Cheng H, Williams S, Correa D et al. Glucose-dependent insulinotropic peptide-overexpressing transgenic mice have increased bone mass. Bone 2007; 40: 1352–1360.
Zhong Q, Itokawa T, Sridhar S, Ding KH, Xie D, Kang B et al. Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am J Physiol Endocrinol Metab 2007; 292: E543–E548.
Lynn FC, Thompson SA, Pospisilik JA, Ehses JA, Hinke SA, Pamir N et al. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. Faseb J 2003; 17: 91–93.
Lim GE, Huang GJ, Flora N, LeRoith D, Rhodes CJ, Brubaker PL . Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine L cell. Endocrinology 2009; 150: 580–591.
Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med 1996; 2: 1254–1258.
Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K et al. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 2004; 53: 1326–1335.
Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y et al. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA 1999; 96: 14843–14847.
Pamir N, Lynn FC, Buchan AM, Ehses J, Hinke SA, Pospisilik JA et al. Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am J Physiol Endocrinol Metab 2003; 284: E931–E939.
Preitner F, Ibberson M, Franklin I, Binnert C, Pende M, Gjinovci A et al. Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. J Clin Invest 2004; 113: 635–645.
Nauck M, Stockmann F, Ebert R, Creutzfeldt W . Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29: 46–52.
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W . Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993; 91: 301–307.
Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001; 86: 3717–3723.
Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ . Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 2001; 50: 609–613.
Vilsboll T, Krarup T, Madsbad S, Holst JJ . Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 2002; 45: 1111–1119.
Minamino N, Kangawa K, Matsuo H . Neuromedin U-8 and U-25: novel uterus stimulating and hypertensive peptides identified in porcine spinal cord. Biochem Biophys Res Commun 1985; 130: 1078–1085.
Austin C, Lo G, Nandha KA, Meleagros L, Bloom SR . Cloning and characterization of the cDNA encoding the human neuromedin U (NmU) precursor: NmU expression in the human gastrointestinal tract. J Mol Endocrinol 1995; 14: 157–169.
Austin C, Oka M, Nandha KA, Legon S, Khandan-Nia N, Lo G et al. Distribution and developmental pattern of neuromedin U expression in the rat gastrointestinal tract. J Mol Endocrinol 1994; 12: 257–263.
Augood SJ, Keast JR, Emson PC . Distribution and characterisation of neuromedin U-like immunoreactivity in rat brain and intestine and in guinea pig intestine. Regul Pept 1988; 20: 281–292.
Ballesta J, Carlei F, Bishop AE, Steel JH, Gibson SJ, Fahey M et al. Occurrence and developmental pattern of neuromedin U-immunoreactive nerves in the gastrointestinal tract and brain of the rat. Neuroscience 1988; 25: 797–816.
Honzawa M, Sudoh T, Minamino N, Kangawa K, Matsuo H . Neuromedin U-like immunoreactivity in rat intestine: regional distribution and immunohistochemical study. Neuropeptides 1990; 15: 1–9.
Kaczmarek P, Malendowicz LK, Pruszynska-Oszmalek E, Wojciechowicz T, Szczepankiewicz D, Szkudelski T et al. Neuromedin U receptor 1 expression in the rat endocrine pancreas and evidence suggesting neuromedin U suppressive effect on insulin secretion from isolated rat pancreatic islets. Int J Mol Med 2006; 18: 951–955.
Alfa RW, Park S, Skelly KR, Poffenberger G, Jain N, Gu X et al. Suppression of insulin production and secretion by a decretin hormone. Cell Metab 2015; 21: 323–333.
Kaczmarek P, Malendowicz LK, Fabis M, Ziolkowska A, Pruszynska-Oszmalek E, Sassek M et al. Does somatostatin confer insulinostatic effects of neuromedin u in the rat pancreas? Pancreas 2009; 38: 208–212.
Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT et al. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 1993; 92: 1349–1356.
Rooman I, Lardon J, Bouwens L . Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 2002; 51: 686–690.
Leung-Theung-Long S, Roulet E, Clerc P, Escrieut C, Marchal-Victorion S, Ritz-Laser B et al. Essential interaction of Egr-1 at an islet-specific response element for basal and gastrin-dependent glucagon gene transactivation in pancreatic alpha-cells. J Biol Chem 2005; 280: 7976–7984.
Ahren B, Pettersson M, Uvnas-Moberg K, Gutniak M, Efendic S . Effects of cholecystokinin (CCK)-8, CCK-33, and gastric inhibitory polypeptide (GIP) on basal and meal-stimulated pancreatic hormone secretion in man. Diabetes Res Clin Pract 1991; 13: 153–161.
Karlsson S, Ahren B . Effects of three different cholecystokinin receptor antagonists on basal and stimulated insulin and glucagon secretion in mice. Acta Physiol Scand 1989; 135: 271–278.
Rushakoff RJ, Goldfine ID, Carter JD, Liddle RA . Physiological concentrations of cholecystokinin stimulate amino acid-induced insulin release in humans. J Clin Endocrinol Metab 1987; 65: 395–401.
Verspohl EJ, Ammon HP . Cholecystokinin (CCK8) regulates glucagon, insulin, and somatostatin secretion from isolated rat pancreatic islets: interaction with glucose. Pflugers Arch 1987; 410: 284–287.
Rushakoff RA, Goldfine ID, Beccaria LJ, Mathur A, Brand RJ, Liddle RA . Reduced postprandial cholecystokinin (CCK) secretion in patients with noninsulin-dependent diabetes mellitus: evidence for a role for CCK in regulating postprandial hyperglycemia. J Clin Endocrinol Metab 1993; 76: 489–493.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011; 6: e25792.
Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 2011; 5: 82–91.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI . Microbial ecology: human gut microbes associated with obesity. Nature 2006; 444: 1022–1023.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. A core gut microbiome in obese and lean twins. Nature 2009; 457: 480–484.
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013; 498: 99–103.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010; 5: e9085.
Moreno-Indias I, Cardona F, Tinahones FJ, Queipo-Ortuno MI . Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol 2014; 5: 190.
Hwang I, Park YJ, Kim YR, Kim YN, Ka S, Lee HY et al. Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J 2015; 29: 2397–2411.
Simon MC, Strassburger K, Nowotny B, Kolb H, Nowotny P, Burkart V et al. Intake of Lactobacillus reuteri improves incretin and insulin secretion in glucose-tolerant humans: a proof of concept. Diabetes Care 2015; 38: 1827–1834.
Cani PD, Lecourt E, Dewulf EM, Sohet FM, Pachikian BD, Naslain D et al. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr 2009; 90: 1236–1243.
Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J 2008; 22: 2416–2426.
Kieffer TJ, Habener JF . The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am J Physiol Endocrinol Metab 2000; 278: E1–E14.
Elmquist JK, Elias CF, Saper CB . From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 1999; 22: 221–232.
Kieffer TJ, Heller RS, Habener JF . Leptin receptors expressed on pancreatic beta-cells. Biochem Biophys Res Commun 1996; 224: 522–527.
Emilsson V, Liu YL, Cawthorne MA, Morton NM, Davenport M . Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes 1997; 46: 313–316.
Kulkarni RN, Wang ZL, Wang RM, Hurley JD, Smith DM, Ghatei MA et al. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest 1997; 100: 2729–2736.
Cases JA, Gabriely I, Ma XH, Yang XM, Michaeli T, Fleischer N et al. Physiological increase in plasma leptin markedly inhibits insulin secretion in vivo. Diabetes 2001; 50: 348–352.
Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF . Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 1997; 46: 1087–1093.
Seufert J, Kieffer TJ, Habener JF . Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc Natl Acad Sci USA 1999; 96: 674–679.
Seufert J, Kieffer TJ, Leech CA, Holz GG, Moritz W, Ricordi C et al. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: implications for the development of adipogenic diabetes mellitus. J Clin Endocrinol Metab 1999; 84: 670–676.
Bradley RL, Cheatham B . Regulation of ob gene expression and leptin secretion by insulin and dexamethasone in rat adipocytes. Diabetes 1999; 48: 272–278.
Kolaczynski JW, Nyce MR, Considine RV, Boden G, Nolan JJ, Henry R et al. Acute and chronic effects of insulin on leptin production in humans: Studies in vivo and in vitro. Diabetes 1996; 45: 699–701.
Zeigerer A, Rodeheffer MS, McGraw TE, Friedman JM . Insulin regulates leptin secretion from 3T3-L1 adipocytes by a PI 3 kinase independent mechanism. Exp Cell Res 2008; 314: 2249–2256.
Barr VA, Malide D, Zarnowski MJ, Taylor SI, Cushman SW . Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology 1997; 138: 4463–4472.
Sattar AA, Sattar R . Insulin-regulated expression of adiponectin receptors in muscle and fat cells. Cell Biol Int 2012; 36: 1293–1297.
Tsuchida A, Yamauchi T, Ito Y, Hada Y, Maki T, Takekawa S et al. Insulin/Foxo1 pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J Biol Chem 2004; 279: 30817–30822.
Cong L, Chen K, Li J, Gao P, Li Q, Mi S et al. Regulation of adiponectin and leptin secretion and expression by insulin through a PI3K-PDE3B dependent mechanism in rat primary adipocytes. Biochem J 2007; 403: 519–525.
Liu BH, Wang YC, Wu SC, Mersmann HJ, Cheng WT, Ding ST . Insulin regulates the expression of adiponectin and adiponectin receptors in porcine adipocytes. Domest Anim Endocrinol 2008; 34: 352–359.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002; 8: 1288–1295.
Wijesekara N, Krishnamurthy M, Bhattacharjee A, Suhail A, Sweeney G, Wheeler MB . Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem 2010; 285: 33623–33631.
Gu W, Li X, Liu C, Yang J, Ye L, Tang J et al. Globular adiponectin augments insulin secretion from pancreatic islet beta cells at high glucose concentrations. Endocrine 2006; 30: 217–221.
Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005; 146: 1764–1771.
Wei L, Hou X, Tatemoto K . Regulation of apelin mRNA expression by insulin and glucocorticoids in mouse 3T3-L1 adipocytes. Regul Pept 2005; 132: 27–32.
Takahashi M, Okimura Y, Iguchi G, Nishizawa H, Yamamoto M, Suda K et al. Chemerin regulates beta-cell function in mice. Sci Rep 2011; 1: 123.
Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Kitazawa R et al. Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett 2008; 582: 573–578.
Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A et al. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes 2009; 58: 2731–2740.
Tan BK, Adya R, Farhatullah S, Lewandowski KC, O'Hare P, Lehnert H et al. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes 2008; 57: 801–808.
Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab 2006; 290: E1253–E1261.
McTernan PG, Fisher FM, Valsamakis G, Chetty R, Harte A, McTernan CL et al. Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes. J Clin Endocrinol Metab 2003; 88: 6098–6106.
Haider DG, Schaller G, Kapiotis S, Maier C, Luger A, Wolzt M . The release of the adipocytokine visfatin is regulated by glucose and insulin. Diabetologia 2006; 49: 1909–1914.
Brown JE, Onyango DJ, Ramanjaneya M, Conner AC, Patel ST, Dunmore SJ et al. Visfatin regulates insulin secretion, insulin receptor signalling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J Mol Endocrinol 2010; 44: 171–178.
Rabe K, Lehrke M, Parhofer KG, Broedl UC . Adipokines and insulin resistance. Mol Med 2008; 14: 741–751.
Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA 2009; 106: 10853–10858.
Ribas F, Villarroya J, Hondares E, Giralt M, Villarroya F . FGF21 expression and release in muscle cells: involvement of MyoD and regulation by mitochondria-driven signalling. Biochem J 2014; 463: 191–199.
Hojman P, Pedersen M, Nielsen AR, Krogh-Madsen R, Yfanti C, Akerstrom T et al. Fibroblast growth factor-21 is induced in human skeletal muscles by hyperinsulinemia. Diabetes 2009; 58: 2797–2801.
Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K . FGF21 is an Akt-regulated myokine. FEBS Lett 2008; 582: 3805–3810.
Trayhurn P, Drevon CA, Eckel J . Secreted proteins from adipose tissue and skeletal muscle - adipokines, myokines and adipose/muscle cross-talk. Arch Physiol Biochem 2011; 117: 47–56.
Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci USA 2008; 105: 13163–13168.
Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 2011; 17: 1481–1489.
Meinert CL, Knatterud GL, Prout TE, Klimt CR . A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. II. Mortality results. Diabetes 1970; 19 (Suppl): 789–830.
Schwartz TB, Meinert CL . The UGDP controversy: thirty-four years of contentious ambiguity laid to rest. Perspect Biol Med 2004; 47: 564–574.
Babenko AP, Gonzalez G, Bryan J . The tolbutamide site of SUR1 and a mechanism for its functional coupling to K(ATP) channel closure. FEBS Lett 1999; 459: 367–376.
Koster JC, Sha Q, Nichols CG . Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol 1999; 114: 203–213.
Clement JPt 4th, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L et al. Association and stoichiometry of K(ATP) channel subunits. Neuron 1997; 18: 827–838.
Miki T, Nagashima K, Seino S . The structure and function of the ATP-sensitive K+ channel in insulin-secreting pancreatic beta-cells. J Mol Endocrinol 1999; 22: 113–123.
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003; 52: 568–572.
Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J et al. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 1996; 16: 1011–1017.
Isomoto S, Kondo C, Yamada M, Matsumoto S, Higashiguchi O, Horio Y et al. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J Biol Chem 1996; 271: 24321–24324.
Proks P, Reimann F, Green N, Gribble F, Ashcroft F . Sulfonylurea stimulation of insulin secretion. Diabetes 2002; 51 (Suppl 3): S368–S376.
Gros L, Virsolvy A, Salazar G, Bataille D, Blache P . Characterization of low-affinity binding sites for glibenclamide on the Kir6.2 subunit of the beta-cell KATP channel. Biochem Biophys Res Commun 1999; 257: 766–770.
Gribble FM, Tucker SJ, Ashcroft FM . The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol 1997; 504 (Pt 1): 35–45.
Overkamp D, Volk A, Maerker E, Heide PE, Wahl HG, Rett K et al. Acute effect of glimepiride on insulin-stimulated glucose metabolism in glucose-tolerant insulin-resistant offspring of patients with type 2 diabetes. Diabetes Care 2002; 25: 2065–2073.
Best JD, Judzewitsch RG, Pfeifer MA, Beard JC, Halter JB, Porte D Jr . The effect of chronic sulfonylurea therapy on hepatic glucose production in non-insulin-dependent diabetes. Diabetes 1982; 31 (4 Pt 1): 333–338.
Kolterman OG, Gray RS, Shapiro G, Scarlett JA, Griffin J, Olefsky JM . The acute and chronic effects of sulfonylurea therapy in type II diabetic subjects. Diabetes 1984; 33: 346–354.
Craig TJ, Ashcroft FM, Proks P . How ATP inhibits the open K(ATP) channel. J Gen Physiol 2008; 132: 131–144.
Markworth E, Schwanstecher C, Schwanstecher M . ATP4- mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes 2000; 49: 1413–1418.
Gangji AS, Cukierman T, Gerstein HC, Goldsmith CH, Clase CM . A systematic review and meta-analysis of hypoglycemia and cardiovascular events: a comparison of glyburide with other secretagogues and with insulin. Diabetes Care 2007; 30: 389–394.
van Staa T, Abenhaim L, Monette J . Rates of hypoglycemia in users of sulfonylureas. J Clin Epidemiol 1997; 50: 735–741.
Ahren B . Avoiding hypoglycemia: a key to success for glucose-lowering therapy in type 2 diabetes. Vasc Health Risk Manag 2013; 9: 155–163.
Fuhlendorff J, Rorsman P, Kofod H, Brand CL, Rolin B, MacKay P et al. Stimulation of insulin release by repaglinide and glibenclamide involves both common and distinct processes. Diabetes 1998; 47: 345–351.
Gribble FM, Tucker SJ, Seino S, Ashcroft FM . Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell K(ATP) channels. Diabetes 1998; 47: 1412–1418.
Hu S, Wang S, Dunning BE . Tissue selectivity of antidiabetic agent nateglinide: study on cardiovascular and beta-cell K(ATP) channels. J Pharmacol Exp Ther 1999; 291: 1372–1379.
Hu S, Wang S, Fanelli B, Bell PA, Dunning BE, Geisse S et al. Pancreatic beta-cell K(ATP) channel activity and membrane-binding studies with nateglinide: a comparison with sulfonylureas and repaglinide. J Pharmacol Exp Ther 2000; 293: 444–452.
Seto Y, Fujita H, Dan K, Fujita T, Kato R . Stimulating activity of A-4166 on insulin release in in situ hamster pancreatic perfusion. Pharmacology 1995; 51: 245–253.
Ikenoue T, Akiyoshi M, Fujitani S, Okazaki K, Kondo N, Maki T . Hypoglycaemic and insulinotropic effects of a novel oral antidiabetic agent, (-)-N-(trans-4-isopropylcyclohexanecarbonyl)-D-phenylalanine (A-4166). Br J Pharmacol 1997; 120: 137–145.
Melander A . Kinetics-effect relations of insulin-releasing drugs in patients with type 2 diabetes: brief overview. Diabetes 2004; 53 (Suppl 3): S151–S155.
Van Gaal LF, Van Acker KL, De Leeuw IH . Repaglinide improves blood glucose control in sulphonylurea-naive type 2 diabetes. Diabetes Res Clin Pract 2001; 53: 141–148.
Furlong NJ, Hulme SA, O'Brien SV, Hardy KJ . Repaglinide versus metformin in combination with bedtime NPH insulin in patients with type 2 diabetes established on insulin/metformin combination therapy. Diabetes Care 2002; 25: 1685–1690.
Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D et al. Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes 2004; 53: 654–662.
Green BD, Gault VA, O'Harte FP, Flatt PR . Structurally modified analogues of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) as future antidiabetic agents. Curr Pharm Des 2004; 10: 3651–3662.
Green BD, Lavery KS, Irwin N, O'Harte FP, Harriott P, Greer B et al. Novel glucagon-like peptide-1 (GLP-1) analog (Val8)GLP-1 results in significant improvements of glucose tolerance and pancreatic beta-cell function after 3-week daily administration in obese diabetic (ob/ob) mice. J Pharmacol Exp Ther 2006; 318: 914–921.
Green BD, Mooney MH, Gault VA, Irwin N, Bailey CJ, Harriott P et al. N-terminal His(7)-modification of glucagon-like peptide-1(7-36) amide generates dipeptidyl peptidase IV-stable analogues with potent antihyperglycaemic activity. J Endocrinol 2004; 180: 379–388.
Irwin N, Gault VA, Green BD, Greer B, Harriott P, Bailey CJ et al. Antidiabetic potential of two novel fatty acid derivatised, N-terminally modified analogues of glucose-dependent insulinotropic polypeptide (GIP): N-AcGIP(LysPAL16) and N-AcGIP(LysPAL37). Biol Chem 2005; 386: 679–687.
Irwin N, Green BD, Mooney MH, Greer B, Harriott P, Bailey CJ et al. A novel, long-acting agonist of glucose-dependent insulinotropic polypeptide suitable for once-daily administration in type 2 diabetes. J Pharmacol Exp Ther 2005; 314: 1187–1194.
Siegel EG, Gallwitz B, Scharf G, Mentlein R, Morys-Wortmann C, Folsch UR et al. Biological activity of GLP-1-analogues with N-terminal modifications. Regul Pept 1999; 79: 93–102.
Ratner RE, Maggs D, Nielsen LL, Stonehouse AH, Poon T, Zhang B et al. Long-term effects of exenatide therapy over 82 weeks on glycaemic control and weight in over-weight metformin-treated patients with type 2 diabetes mellitus. Diabetes Obes Metab 2006; 8: 419–428.
Moretto TJ, Milton DR, Ridge TD, Macconell LA, Okerson T, Wolka AM et al. Efficacy and tolerability of exenatide monotherapy over 24 weeks in antidiabetic drug-naive patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2008; 30: 1448–1460.
Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28: 1083–1091.
Buse JB, Drucker DJ, Taylor KL, Kim T, Walsh B, Hu H et al. DURATION-1: exenatide once weekly produces sustained glycemic control and weight loss over 52 weeks. Diabetes Care 2010; 33: 1255–1261.
Kim D, MacConell L, Zhuang D, Kothare PA, Trautmann M, Fineman M et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007; 30: 1487–1493.
Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 2009; 374: 39–47.
Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P et al. Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD). Diabetes Care 2009; 32: 1224–1230.
Fonseca VA, Alvarado-Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE . Efficacy and safety of the once-daily GLP-1 receptor agonist lixisenatide in monotherapy: a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes (GetGoal-Mono). Diabetes Care 2012; 35: 1225–1231.
Ratner RE, Rosenstock J, Boka G . Dose-dependent effects of the once-daily GLP-1 receptor agonist lixisenatide in patients with Type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled trial. Diabet Med 2010; 27: 1024–1032.
Rosenstock J, Hanefeld M, Shamanna P, Min KW, Boka G, Miossec P et al. Beneficial effects of once-daily lixisenatide on overall and postprandial glycemic levels without significant excess of hypoglycemia in type 2 diabetes inadequately controlled on a sulfonylurea with or without metformin (GetGoal-S). J Diabetes Complications 2014; 28: 386–392.
Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL et al. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7-37) in normal and diabetic subjects. Regul Pept 1994; 51: 63–74.
Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS et al. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes 2009; 58: 1342–1349.
Mentis N, Vardarli I, Kothe LD, Holst JJ, Deacon CF, Theodorakis M et al. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 2011; 60: 1270–1276.
Herrmann C, Goke R, Richter G, Fehmann HC, Arnold R, Goke B . Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion 1995; 56: 117–126.
Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF . Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992; 15: 270–276.
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 854–865.
Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000; 49: 2063–2069.
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 2002; 51: 2074–2081.
Seifarth C, Schehler B, Schneider HJ . Effectiveness of metformin on weight loss in non-diabetic individuals with obesity. Exp Clin Endocrinol Diabetes 2013; 121: 27–31.
Lee A, Morley JE . Metformin decreases food consumption and induces weight loss in subjects with obesity with type II non-insulin-dependent diabetes. Obes Res 1998; 6: 47–53.
Hashemitabar M, Bahramzadeh S, Saremy S, Nejaddehbashi F . Glucose plus metformin compared with glucose alone on beta-cell function in mouse pancreatic islets. Biomed Rep 2015; 3: 721–725.
Richardson H, Campbell SC, Smith SA, Macfarlane WM . Effects of rosiglitazone and metformin on pancreatic beta cell gene expression. Diabetologia 2006; 49: 685–696.
Jiang Y, Huang W, Wang J, Xu Z, He J, Lin X et al. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci 2014; 10: 268–277.
Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H et al. Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol 2004; 68: 409–416.
Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab 2004; 89: 5535–5541.
Lupi R, Del Guerra S, Fierabracci V, Marselli L, Novelli M, Patane G et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 2002; 51 (Suppl 1): S134–S137.
Murphy EJ, Davern TJ, Shakil AO, Shick L, Masharani U, Chow H et al. Troglitazone-induced fulminant hepatic failure. Acute Liver Failure Study Group. Dig Dis Sci 2000; 45: 549–553.
Neuschwander-Tetri BA, Isley WL, Oki JC, Ramrakhiani S, Quiason SG, Phillips NJ et al. Troglitazone-induced hepatic failure leading to liver transplantation. A case report. Ann Intern Med 1998; 129: 38–41.
Kohlroser J, Mathai J, Reichheld J, Banner BF, Bonkovsky HL . Hepatotoxicity due to troglitazone: report of two cases and review of adverse events reported to the United States Food and Drug Administration. Am J Gastroenterol 2000; 95: 272–276.
Nissen SE, Wolski K . Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007; 356: 2457–2471.
Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS et al. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 1996; 137: 4189–4195.
Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S et al. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 1996; 15: 5336–5348.
Dumasia R, Eagle KA, Kline-Rogers E, May N, Cho L, Mukherjee D . Role of PPAR- gamma agonist thiazolidinediones in treatment of pre-diabetic and diabetic individuals: a cardiovascular perspective. Curr Drug Targets Cardiovasc Haematol Disord 2005; 5: 377–386.
Smith SR, De Jonge L, Volaufova J, Li Y, Xie H, Bray GA . Effect of pioglitazone on body composition and energy expenditure: a randomized controlled trial. Metabolism 2005; 54: 24–32.
Ishida H, Takizawa M, Ozawa S, Nakamichi Y, Yamaguchi S, Katsuta H et al. Pioglitazone improves insulin secretory capacity and prevents the loss of beta-cell mass in obese diabetic db/db mice: Possible protection of beta cells from oxidative stress. Metabolism 2004; 53: 488–494.
Diani AR, Sawada G, Wyse B, Murray FT, Khan M . Pioglitazone preserves pancreatic islet structure and insulin secretory function in three murine models of type 2 diabetes. Am J Physiol Endocrinol Metab 2004; 286: E116–E122.
Kawasaki F, Matsuda M, Kanda Y, Inoue H, Kaku K . Structural and functional analysis of pancreatic islets preserved by pioglitazone in db/db mice. Am J Physiol Endocrinol Metab 2005; 288: E510–E518.
Kimura T, Kaneto H, Shimoda M, Hirukawa H, Okauchi S, Kohara K et al. Protective effects of pioglitazone and/or liraglutide on pancreatic beta-cells in db/db mice: comparison of their effects between in an early and advanced stage of diabetes. Mol Cell Endocrinol 2015; 400: 78–89.
Kanda Y, Shimoda M, Hamamoto S, Tawaramoto K, Kawasaki F, Hashiramoto M et al. Molecular mechanism by which pioglitazone preserves pancreatic beta-cells in obese diabetic mice: evidence for acute and chronic actions as a PPARgamma agonist. Am J Physiol Endocrinol Metab 2010; 298: E278–E286.
Wachters-Hagedoorn RE, Priebe MG, Heimweg JA, Heiner AM, Elzinga H, Stellaard F et al. Low-dose acarbose does not delay digestion of starch but reduces its bioavailability. Diabet Med 2007; 24: 600–606.
Gerard J, Luyckx AS, Lefebvre PJ . Improvement of metabolic control in insulin dependent diabetics treated with the alpha-glucosidase inhibitor acarbose for two months. Diabetologia 1981; 21: 446–451.
Josse RG, Chiasson JL, Ryan EA, Lau DC, Ross SA, Yale JF et al. Acarbose in the treatment of elderly patients with type 2 diabetes. Diabetes Res Clin Pract 2003; 59: 37–42.
Meneilly GS, Ryan EA, Radziuk J, Lau DC, Yale JF, Morais J et al. Effect of acarbose on insulin sensitivity in elderly patients with diabetes. Diabetes Care 2000; 23: 1162–1167.
Baron AD, Eckel RH, Schmeiser L, Kolterman OG . The effect of short-term alpha-glucosidase inhibition on carbohydrate and lipid metabolism in type II (noninsulin-dependent) diabetics. Metabolism 1987; 36: 409–415.
Hillebrand I, Boehme K, Frank G, Fink H, Berchtold P . The effects of the alpha-glucosidase inhibitor BAY g 5421 (Acarbose) on postprandial blood glucose, serum insulin, and triglyceride levels: dose-time-response relationships in man. Res Exp Med (Berl) 1979; 175: 87–94.
Wolever TM, Chiasson JL, Josse RG, Hunt JA, Palmason C, Rodger NW et al. Small weight loss on long-term acarbose therapy with no change in dietary pattern or nutrient intake of individuals with non-insulin-dependent diabetes. Int J Obes Relat Metab Disord 1997; 21: 756–763.
Nakhaee A, Sanjari M . Evaluation of effect of acarbose consumption on weight losing in non-diabetic overweight or obese patients in Kerman. J Res Med Sci 2013; 18: 391–394.
Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M . Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance: the STOP-NIDDM trial. JAMA 2003; 290: 486–494.
Goda T, Suruga K, Komori A, Kuranuki S, Mochizuki K, Makita Y et al. Effects of miglitol, an alpha-glucosidase inhibitor, on glycaemic status and histopathological changes in islets in non-obese, non-insulin-dependent diabetic Goto-Kakizaki rats. Br J Nutr 2007; 98: 702–710.
Koyama M, Wada R, Mizukami H, Sakuraba H, Odaka H, Ikeda H et al. Inhibition of progressive reduction of islet beta-cell mass in spontaneously diabetic Goto-Kakizaki rats by alpha-glucosidase inhibitor. Metabolism 2000; 49: 347–352.
Fukaya N, Mochizuki K, Tanaka Y, Kumazawa T, Jiuxin Z, Fuchigami M et al. The alpha-glucosidase inhibitor miglitol delays the development of diabetes and dysfunctional insulin secretion in pancreatic beta-cells in OLETF rats. Eur J Pharmacol 2009; 624: 51–57.
Hanefeld M, Schaper F . Acarbose: oral anti-diabetes drug with additional cardiovascular benefits. Expert Rev Cardiovasc Ther 2008; 6: 153–163.
Hussain MA, Daniel PB, Habener JF . Glucagon stimulates expression of the inducible cAMP early repressor and suppresses insulin gene expression in pancreatic beta-cells. Diabetes 2000; 49: 1681–1690.
Greenbaum CJ, Havel PJ, Taborsky GJ Jr, Klaff LJ . Intra-islet insulin permits glucose to directly suppress pancreatic A cell function. J Clin Invest 1991; 88: 767–773.
Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 2006; 3: 47–58.
Meier JJ, Kjems LL, Veldhuis JD, Lefebvre P, Butler PC . Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 2006; 55: 1051–1056.
Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 2003; 52: 1799–1805.
de Jager J, Dekker JM, Kooy A, Kostense PJ, Nijpels G, Heine RJ et al. Endothelial dysfunction and low-grade inflammation explain much of the excess cardiovascular mortality in individuals with type 2 diabetes: the Hoorn Study. Arterioscler Thromb Vasc Biol 2006; 26: 1086–1093.
Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M . Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339: 229–234.
Almdal T, Scharling H, Jensen JS, Vestergaard H . The independent effect of type 2 diabetes mellitus on ischemic heart disease, stroke, and death: a population-based study of 13,000 men and women with 20 years of follow-up. Arch Intern Med 2004; 164: 1422–1426.
Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR . Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 2003; 63: 225–232.
Ravid M, Brosh D, Ravid-Safran D, Levy Z, Rachmani R . Main risk factors for nephropathy in type 2 diabetes mellitus are plasma cholesterol levels, mean blood pressure, and hyperglycemia. Arch Intern Med 1998; 158: 998–1004.
Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism 2003; 52: 605–608.
Wannamethee SG, Lowe GD, Rumley A, Cherry L, Whincup PH, Sattar N . Adipokines and risk of type 2 diabetes in older men. Diabetes Care 2007; 30: 1200–1205.
Milewicz A, Mikulski E, Bidzinska B . Plasma insulin, cholecystokinin, galanin, neuropeptide Y and leptin levels in obese women with and without type 2 diabetes mellitus. Int J Obes Relat Metab Disord 2000; 24 Suppl 2: S152–S153.
Katsuki A, Urakawa H, Gabazza EC, Murashima S, Nakatani K, Togashi K et al. Circulating levels of active ghrelin is associated with abdominal adiposity, hyperinsulinemia and insulin resistance in patients with type 2 diabetes mellitus. Eur J Endocrinol 2004; 151: 573–577.
Poykko SM, Kellokoski E, Horkko S, Kauma H, Kesaniemi YA, Ukkola O . Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 2003; 52: 2546–2553.
Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001; 344: 1343–1350.
Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R . Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011; 54: 2506–2514.
Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ et al. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci USA 2008; 105: 3992–3997.
Shu Y, Liu X, Yang Y, Takahashi M, Gillis KD . Phosphorylation of SNAP-25 at Ser187 mediates enhancement of exocytosis by a phorbol ester in INS-1 cells. J Neurosci 2008; 28: 21–30.
Lilja L, Johansson JU, Gromada J, Mandic SA, Fried G, Berggren PO et al. Cyclin-dependent kinase 5 associated with p39 promotes Munc18-1 phosphorylation and Ca(2+)-dependent exocytosis. J Biol Chem 2004; 279: 29534–29541.
Dixit SS, Wang T, Manzano EJ, Yoo S, Lee J, Chiang DY et al. Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PLoS ONE 2013; 8: e58655.
Dzhura I, Chepurny OG, Leech CA, Roe MW, Dzhura E, Xu X et al. Phospholipase C-epsilon links Epac2 activation to the potentiation of glucose-stimulated insulin secretion from mouse islets of Langerhans. Islets 2011; 3: 121–128.
Kang L, He Z, Xu P, Fan J, Betz A, Brose N et al. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab 2006; 3: 463–468.
Kwan EP, Xie L, Sheu L, Nolan CJ, Prentki M, Betz A et al. Munc13-1 deficiency reduces insulin secretion and causes abnormal glucose tolerance. Diabetes 2006; 55: 1421–1429.
Yaekura K, Julyan R, Wicksteed BL, Hays LB, Alarcon C, Sommers S et al. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem 2003; 278: 9715–9721.
Torii S, Takeuchi T, Nagamatsu S, Izumi T . Rab27 effector granuphilin promotes the plasma membrane targeting of insulin granules via interaction with syntaxin 1a. J Biol Chem 2004; 279: 22532–22538.
Betts GJ, Desaix P, Johnson E, Korol O, Kruse D, Poe B et al. Human Anatomy and Physiology. OpenStax College: Houston, TX, USA, 2013.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Röder, P., Wu, B., Liu, Y. et al. Pancreatic regulation of glucose homeostasis. Exp Mol Med 48, e219 (2016). https://doi.org/10.1038/emm.2016.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.6
This article is cited by
-
Role of cytosolic and endoplasmic reticulum Ca2+ in pancreatic beta-cells: pros and cons
Pflügers Archiv - European Journal of Physiology (2024)
-
The potential of therapeutic strategies targeting mitochondrial biogenesis for the treatment of insulin resistance and type 2 diabetes mellitus
Archives of Pharmacal Research (2024)
-
Small interfering RNA (siRNA) as a potential gene silencing strategy for diabetes and associated complications: challenges and future perspectives
Journal of Diabetes & Metabolic Disorders (2024)
-
Transient loss of consciousness immediately after total pancreatectomy for pancreatic metastases from renal cell carcinoma: a case report
Surgical Case Reports (2023)
-
Dillenia indica fruit extract alleviates sucrose-induced fatty liver and improves serum biochemical alterations in mice
Nutrire (2023)
